A new spin (label) on Alphafold2: DEERfold guides Alphafold2 modeling with DEER distance distributions
By Shelby A. Harris
 No one can argue that the development of AlphaFold2 (AF2) has been one of the greatest achievements in the realm of protein structure prediction. However, AF2 has been plagued by two major limitations: (1) The quality of the predicted models depends on the quality of the multiple sequence alignment (MSA) input and (2) The assumption that this input sequence only folds into one conformation. It is known that proteins are dynamic models that have multiple folding states in order to carry out their biological function.
No one can argue that the development of AlphaFold2 (AF2) has been one of the greatest achievements in the realm of protein structure prediction. However, AF2 has been plagued by two major limitations: (1) The quality of the predicted models depends on the quality of the multiple sequence alignment (MSA) input and (2) The assumption that this input sequence only folds into one conformation. It is known that proteins are dynamic models that have multiple folding states in order to carry out their biological function.
Although these limitations exist, there has always been a desire to refine and guide AF2 with experimental data, especially in the context of incorporating the protein energy landscape. While there have been methods to “hack” AF2, performance of the algorithm has been variable. Some tools, AlphaLink which uses cross-linking mass spectrometry data, have been developed and demonstrate the ability to tune AF2 predictions with collected data. This proves that AF2 has the flexibility to accept these inputs, but predictions of multiple conformations have yet to be addressed. This is largely due to the fact that data collected from probe-based spectroscopic techniques, such as Double Electron Electron Resonance (DEER) spectroscopy, present a unique challenge due to the probe’s flexible movement, or rotameric freedom, relative to the protein backbone.
To tackle this issue, members of the Benjamin Brown lab and the Mchaourab lab set out to develop an AF2 tool that can incorporate DEER distributions and provide a blueprint for other spatial constraints.
Dubbed “DEERFold,” this new tool was created by fine-tuning AF2 within the OpenFold framework to interpret and integrate spin-label distance distributions from DEER spectroscopy directly into the network architecture. While AF2 is limited by its training on the Protein Data Bank (PDB) and typically yields a single structure, DEERFold can predict alternative, heterogeneous conformational ensembles when distance constraints are applied. A systematic evaluation showed that 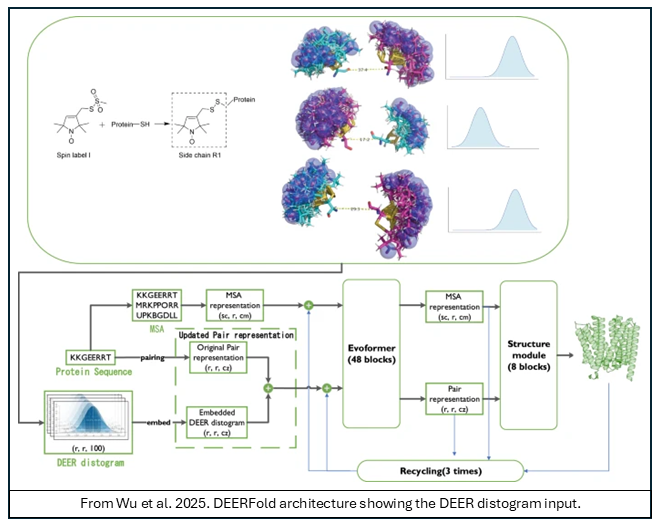 incorporating either experimental or simulated DEER constraints successfully drives the model toward the desired conformation. Importantly, the authors found that the content of the constraints, rather than their exact shape, is the main factor in improving the quality of the predictions. These findings highlight DEERFold’s utility in modeling conformational changes, even when initial or target experimental structures are not known.
incorporating either experimental or simulated DEER constraints successfully drives the model toward the desired conformation. Importantly, the authors found that the content of the constraints, rather than their exact shape, is the main factor in improving the quality of the predictions. These findings highlight DEERFold’s utility in modeling conformational changes, even when initial or target experimental structures are not known.
DEERFold’s success shows that AF2 can be molded to include many other forms of experimental data, such as nuclear magnetic resonance (NMR), fluorescence resonance energy transfer (FRET), hydrogen-deuterium exchange (HDX), and cross-link mass spectrometry (XL-MS).
The authors hope that DEERFold could be used as a platform for this inclusion and bring in extremely valuable data to enhance protein structure prediction.
Read the full paper in Nature Communications.
Leave a Response
You must be logged in to post a comment