Liu, Yunchao; Moretti, Rocco; Wang, Yu; Dong, Ha; Yan, Bailu; Bodenheimer, Bobby; Derr, Tyler; Meiler, Jens. “Advancements in ligand-based virtual screening through the synergistic integration of graph neural networks and expert-crafted descriptors.” Journal of Chemical Information and Modeling 65, no. 10 (2025): 4898-4905. https://doi.org/10.1021/acs.jcim.5c00822.
Combining traditional chemical information (called “descriptors”) with advanced machine learning models known as graph neural networks (GNNs) offers a promising way to improve how scientists screen potential drug molecules—a process known as ligand-based virtual screening. In this study, researchers tested how well different types of GNNs benefited from this combination.
They found that some models, like GCN and SchNet, improved a lot when descriptors were added, while another model, SphereNet, only improved slightly. Still, all three models—GCN, SchNet, and SphereNet—performed about the same when this combination approach was used. This suggests that even simpler GNNs can work just as well as more complex ones if you add the right chemical information.
The researchers also discovered that a set of expert-designed descriptors performed very well on their own, especially in challenging tests that mimic real-world drug discovery. In fact, these descriptors sometimes did better than the GNN-descriptor combinations. This highlights the need for new GNN models that can handle these real-world challenges more effectively.
Overall, this study shows that blending chemical knowledge with machine learning can improve virtual drug screening and points to new directions for improving these tools in the future. The code for this work is available at https://github.com/meilerlab/gnn-descriptor.
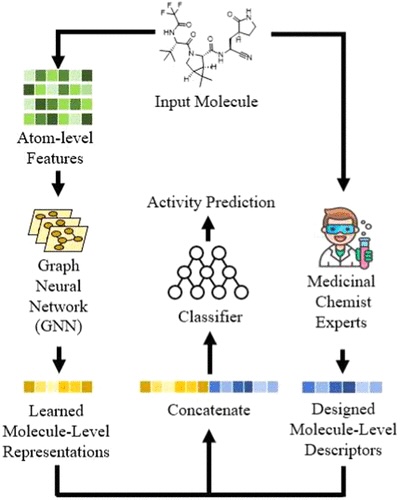
Figure 1. Overview of the investigated method. The learned molecular representation of GNN is concatenated with expert-crafted descriptors to enhance the predictive power.