Luo, Can; Peters, Brock A.; Zhou, Xin Maizie. “Large indel detection in region-based phased diploid assemblies from linked-reads.” BMC Genomics 26, no. Suppl 2 (2025): 263. https://doi.org/10.1186/s12864-025-11398-z.
Scientists study our DNA to understand genetic differences that may affect health or traits. One way to do this is by using a method called “linked-reads,” which helps piece together long stretches of DNA and find changes in the genome. However, analyzing whole genomes with linked-reads usually requires a lot of computer power, which can be a problem for large-scale studies.
To address this, we developed a new method called RegionIndel. Instead of looking at the entire genome, RegionIndel focuses only on specific smaller sections (usually 50,000 DNA letters at a time). It uses barcodes to group DNA reads from the same region, then puts the pieces together while keeping track of which parent each piece came from. Finally, it compares the assembled sequences to a reference to find large insertions and deletions—types of genetic changes known as structural variants (SVs).
We tested RegionIndel on a well-known human DNA sample called HG002, using two different types of linked-read data. RegionIndel found more structural variants—and did so more accurately—than several other existing methods. For example, when analyzing one type of data (called 10x linked-reads), it correctly identified about 75% of deletions and 62% of insertions. It also did a good job figuring out whether someone had 0, 1, or 2 copies of a variant, with accuracy over 80%.
Figure 1
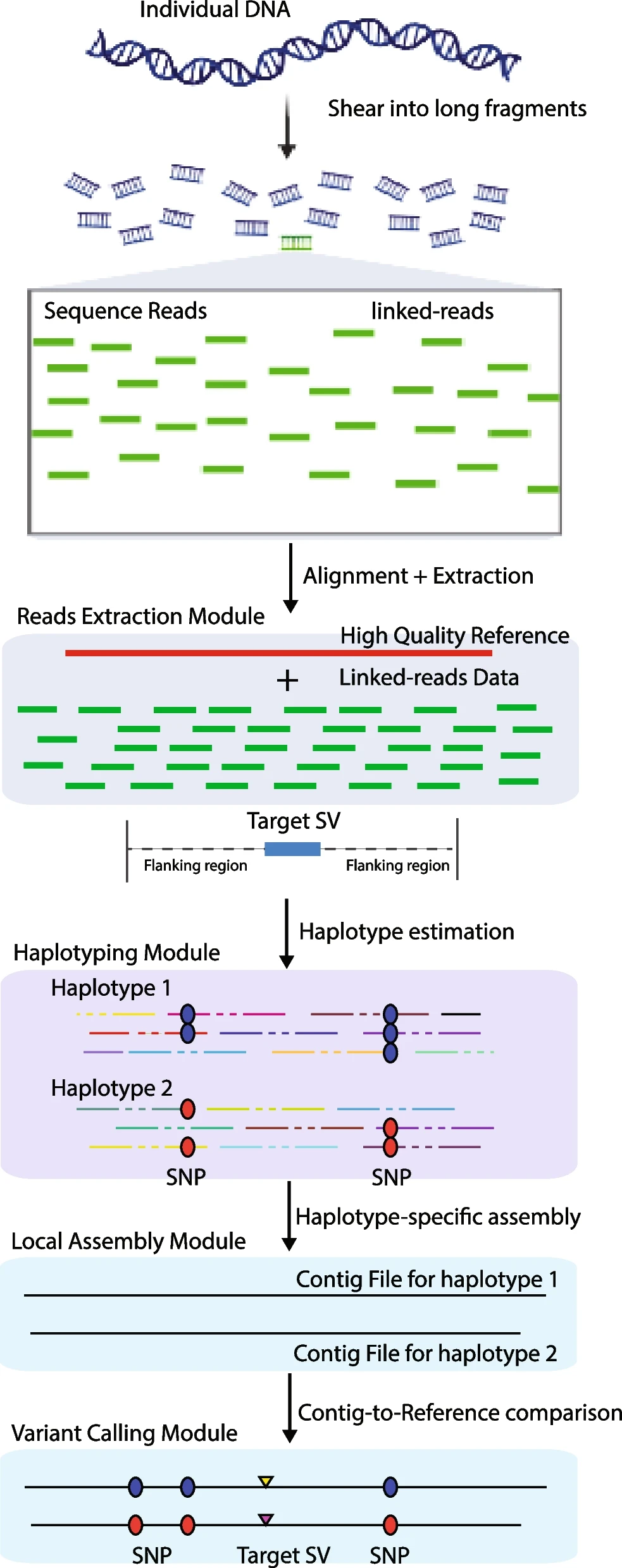
Schematic diagram of the RegionIndel pipeline. Input data are the high-quality reference genome and barcoded reads, each with a barcode (not shown). The reads extraction module extracts barcoded reads aligning to the region of interest. The reads with the same barcode (read cloud) form the virtual long fragment molecule. The haplotyping module partitions molecules into different parental haplotypes, and then the corresponding barcoded reads will be partitioned into different haplotypes. The local assembly module takes barcoded reads and performs de novo local assembly independently for each haplotype through SPAdes. Finally, the variant calling module integrates paftools to perform a contig-to-reference comparison to detect all variants