| |
|
|
|
|

Our group studies ultrafast carrier relaxation dynamics in semiconducting nanocrystals and polymers as well as solvation dynamics of important proteins using the technique of femtosecond fluorescence upconversion.
Ultrafast spectroscopic investigations of carrier dephasing and relaxation in monodisperse, defect-free semiconducting nanocrystals have been performed by very few groups. In each case, high-quality nanocrystals were supplied by Alivisatos. The important distinction between these nanocrystals and nanoclusters prepared by different methods is that the interior of high-quality nanocrystals is truly molecular. The position and constitution of each atom is known and there are no interior vacancies which act as traps for carriers. The emission spectrum of nanoclusters has little band-edge emission and is dominated by a broad, weak, red-shifted emission feature.
Pump-probe data previously obtained by Prof. Rosenthal during her work with Chuck Shank on these materials shows two decay components: a short, 80 fs component with a ligand-dependent amplitude and a longer, poorly determined, picosecond component. The short time decay was attributed to electrons localizing to surface cadmium atoms, whereas the longer component was attributed to localization of the hole on surface selenium atoms.
One of our goals is to determine the carrier migration rate from the donor nanocrystal to both electron and hole acceptors. In order to determine this, it is essential to first unambiguously determine the electron and hole trapping rates. Femtosecond fluorescence upconversion is the ideal technique for this problem, since pump-probe transients intrinsically contain contributions from excited state absorption, stimulated emission, and ground state recovery, thus the kinetics would be obfuscated.
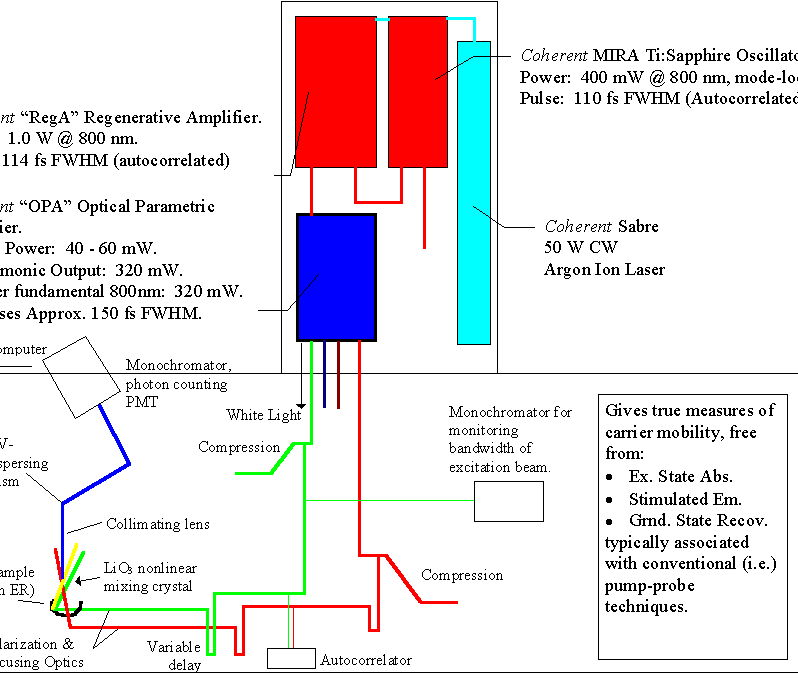
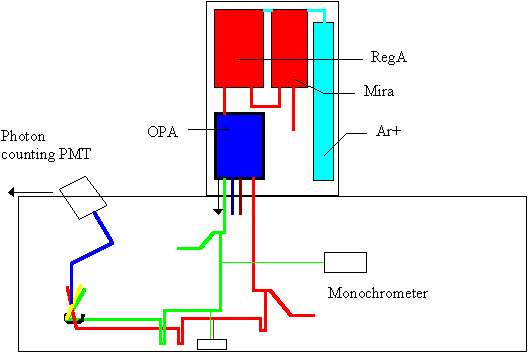
Our ultrafast laser system consists of (all Coherent, Inc.) a Sabre argon ion pump source, which drives a Ti:Sapphire oscillator (Mira 900) and a regenerative amplifier (RegA 9000). The 80 MHz output of the Mira contains 90 fs pulses with nanojoule energies per pulse, centered at 794 nm. A portion of this output is fed into the RegA, where amplification of the Mira pulse leads to microjoule energies per pulse at a repetition rate of 250 kHz. The resulting pulse is re-compressed to approximately 180 fs after amplification, which is short and energetic enough to produce self-focusing in almost any medium to generate a white-light continuum. We use an optical parametric amplifier (OPA) as the laser sources of our experiments. The output of the OPA provides a plethora of wavelengths: 300 mW @ 400nm, 30 mW from 480-760nm, 200 mW @ 794 (residual pump source), 50 mW of white light, and finally an idler beam which extends from 940 - 2400nm. The pulses are recompressed externally with a standard prism pair.
A schematic of our setup is shown below. (To see a larger, more descriptive version, click here.) For the femtosecond fluorescence upconversion, the tunable visible beam is used as the excitation source, and the residual 794 nm pump source for the OPA is used as the gate.
{kind=link}
The concept of the experiment is quite simple, but the actual implementation proves to be quite difficult. Basically, the phenomenon of sum frequency generation is employed. When two laser pulses overlap in time and space in a nonlinear medium (a 0.5 mm LiIO3 crystal in our case), a frequency is produced at the sum of the two frequencies: vsum=v1+v2, or:
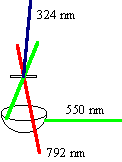 So, a pulse centered at 792 nm overlapping with a pulse at 550 nm produces a sum frequency at 324 nm as shown; this is in fact how our instrument response function is obtained. By scanning one of the pulses along a delay line, the shape of the pulses used to excite the sample are formed, since the intensity of the sum frequency is proportional to the intensities of the two input pulses (the individual pulses are Gaussian in intensity). Selecting a wavelength for upconversion is performed by changing the phase-matching angle of the nonlinear mixing crystal.
So, a pulse centered at 792 nm overlapping with a pulse at 550 nm produces a sum frequency at 324 nm as shown; this is in fact how our instrument response function is obtained. By scanning one of the pulses along a delay line, the shape of the pulses used to excite the sample are formed, since the intensity of the sum frequency is proportional to the intensities of the two input pulses (the individual pulses are Gaussian in intensity). Selecting a wavelength for upconversion is performed by changing the phase-matching angle of the nonlinear mixing crystal.
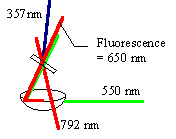
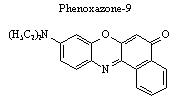 The same process is employed to extract the fluorescence decay profiles of an analyte. In this case, the fluorescence of the sample is mixed with the gate beam, and the sum frequency of the two is detected. By scanning one of the path lengths in time, a fluorescence decay profile is obtained. The example to the left shows the setup for upconverting the laser dye phenoxazone-9 in ethanol. The sample cell is positioned at the foci of an elliptical reflector and the resulting fluorescence is centered at the other foci, approximately 12 cm away in the nonlinear mixing crystal.
The same process is employed to extract the fluorescence decay profiles of an analyte. In this case, the fluorescence of the sample is mixed with the gate beam, and the sum frequency of the two is detected. By scanning one of the path lengths in time, a fluorescence decay profile is obtained. The example to the left shows the setup for upconverting the laser dye phenoxazone-9 in ethanol. The sample cell is positioned at the foci of an elliptical reflector and the resulting fluorescence is centered at the other foci, approximately 12 cm away in the nonlinear mixing crystal.

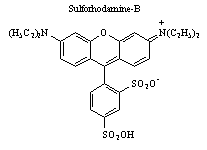 Phenoxazone-9 has a 100 nm Stokes shift, which makes it a relatively easy sample to study. However, when the sample has absorption and emission wavelengths which are close together (such as sulforhodaine-B, Stokes shift = 19 nm), the separation of the cross-correlation signal from that of the fluorescence becomes increasingly more difficult. To overcome this, a UV-dispersing prism and an iris is utilized for resolving a particular wavelength.
Phenoxazone-9 has a 100 nm Stokes shift, which makes it a relatively easy sample to study. However, when the sample has absorption and emission wavelengths which are close together (such as sulforhodaine-B, Stokes shift = 19 nm), the separation of the cross-correlation signal from that of the fluorescence becomes increasingly more difficult. To overcome this, a UV-dispersing prism and an iris is utilized for resolving a particular wavelength.
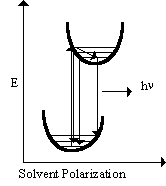 Fluorescence upconversion is particularly suited for studying transient solvation dynamics in polar solvents. The dye molecule Phenoxazone-9 has an excited state dipole moment larger than that in the ground state. When the molecule is excited by the laser pulse, the resulting dipole moment influences the surrounding solvent molecules such that they reorient themselves to accommodate the new dipole moment. If the fluorescence spectrum of the molecule is influenced by the change in surrounding solvent molecules, then the fluorescence spectrum may show a rapid red shift on the timescale of the microscopic solvent reorientation. The potential energy diagram of this process is shown at the right. If this process is to occur, the expected result in the fluorescence decay profile would be (Barbara et al., Rev. Sci. Instrum., 59(7), 1988):
Fluorescence upconversion is particularly suited for studying transient solvation dynamics in polar solvents. The dye molecule Phenoxazone-9 has an excited state dipole moment larger than that in the ground state. When the molecule is excited by the laser pulse, the resulting dipole moment influences the surrounding solvent molecules such that they reorient themselves to accommodate the new dipole moment. If the fluorescence spectrum of the molecule is influenced by the change in surrounding solvent molecules, then the fluorescence spectrum may show a rapid red shift on the timescale of the microscopic solvent reorientation. The potential energy diagram of this process is shown at the right. If this process is to occur, the expected result in the fluorescence decay profile would be (Barbara et al., Rev. Sci. Instrum., 59(7), 1988):
- A rapid decrease in fluorescence intensity after excitation at wavelengths to the blue edge of the emission,
- A rapid rise after excitation at wavelengths to the red side of the fluorescence emission,
- Little or no fast kinetics at wavelengths in the middle of the fluorescence emission.
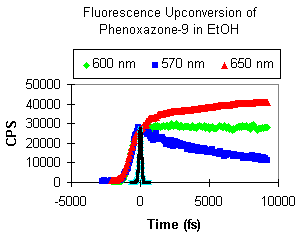 Indeed, we have evidence of this behavior as shown in the wavelength-dependent fluorescence upconversion of Phenoxazone-9 in ethanol. As expected, a rapid decrease in the emission is evident on the blue side, an increase on the red side, and no shift in the middle (fluorescence max = 600 nm). We have seen similar behavior for Sulforhodamine-B and Crysyl Violet laser dyes.
Indeed, we have evidence of this behavior as shown in the wavelength-dependent fluorescence upconversion of Phenoxazone-9 in ethanol. As expected, a rapid decrease in the emission is evident on the blue side, an increase on the red side, and no shift in the middle (fluorescence max = 600 nm). We have seen similar behavior for Sulforhodamine-B and Crysyl Violet laser dyes.
Emission wavelengths from our nanocrystals translates directly into electron and/or hole dynamics. By tuning the nonlinear mixing crystal to upconvert band-edge or red-shifted emission, we are able to decipher carrier mobilities and trapping timescales which are crucial pieces of evidence for our ultimate goals of device manufacture.