Chapter 25
Q: How detailed of an understanding will we need of glycolysis? Should we focus on the steps or simply know what the purpose of glycolysis is and that it involves sugars?
A: The first two steps of the glycolysis pathways is an isomerization of glucose to fructose, followed by a retro-aldol reaction. You should be familiar with that chemistry. You don't need to know anything about the glycolysis (enzymatic) pathway.
Q: I was going through chapter 25 problems, and a lot of them require us to refer to the structures of different carbohydrates. For example, question 3 asks us to draw the Fischer projection for L-(+)-arabinose. Without having committed the structure of arabinose to memory, I needed to go back a page and look at its enantiomer, D-(-)-arabinose. Should we memorize the structures of all the sugars on page1027, or will you provide us with the structures?
A: The only carbohydrate structure that you should know is D-glyceraldehyde. You do know need to know the structures of any of the other carbohydrates.
Q: I am a bit confused about D/L vs. R/S vs (+)/(-). On page 1026 of the book, it states that "Optical activities cannot be determined directly from the D and L prefixes. As it turns out, both D-erythrose and D-threose are levorotatory..." On you slide, you have (+)-rotation=dextrorotatory=D. I understand that there isn't a correlation between D/L and R/S but I was wondering if you might elucidate where (+)/(-) and dextrorotatory/levorotatory fit into the picture.
A: Slide 250 is for D- and L-glyceraldehyde only, from where the D/L nomenclature was derived. For D carbohydrates, the stereochemistry of the highest numbered chiral carbon is D, because it is the same as D-glyceraldehyde. Optically active molecules that rotate plane polarized light in the (+)-direction can be designated as d as well (for dextrorotatory), but with a small case d to distinguish it from the carbohydrate designation. So a (+)-rotation is that same a (d)-rotation and a (-)-rotation is the same as the (l)-rotation (for levorotatory).
Chapter 22
Answer to the combine spectral problem on slide 227
Q: On slide 221, for several of the transformations shown there are 2 reagents listed, such as Cu2O and H2O for the phenol formation. Should we know both of these reagents for the transformations that show 2 or will 1suffice?
A: Some other text books note that CuCl, CuBr, and Cu2O are needed to convert an aryldiazonium ion to an aryl chloride, aryl bromide, and phenol, respectively. This books just uses heat and HCl, HBr, and H2O. If you are asked to provide reagents for such a transformation, both conditions are acceptable. However, if you are given the reagents and asked to provide a product, you should at least be familiar enough with both conditions so that the presence (or absence) of the copper salt won't throw you off.
Q: Friedel-Crafts does not work on aryl amines unless the NH2 group is protected (page 934); however, I remember Professor Sulikowski also mentioning that the Friedel-Crafts does not work on nitrobenzenes either. Is this true or am I remembering incorrectly? In problem 22.18, it shows Br2/Fe working on nitrobenzene. Is halogenation of benezene via Br2/Fe different from Br2/AlBr3? Also, can either work on nitrobenzene?
A: That is correct. The problem is that amino group is a Lewis base and actually reacts with the AlCl3 (a strong Lewis acid) catalyst. Once this acid-base chemistry takes place, there is no F-C reaction. Your memory serves you well, F-C reaction do not occur of highly deactivated rings; in fact, an F-C acylation of say toluene can be done in nitrobenzene as the solvent. So the rule noted in the book is that F-C acylation reaction only take place with an aromatic that is at least as reactive as a halobenzene; acetamides (the amide from protection of anilines) are activating. This rule is limited to the F-C reactions. Other electrophilic aromatic substitution reactions such as halogenations, nitrations, etc, work on nitrobenzenes but the rates are slow.
I think that is a typo in the solutions manuel. 22.18 should be FeBr3, which is equivalent to AlBr3 for electrophilic aromatic substitution reactions.
Q: I was wondering why amide reduction works with LiAlH4, then H2O but not with H2/Pd (catalyst)... I know in the text in section 22.9 it mentions reduction of the carbonyl group of an amide by lithium aluminum hydride.
A: The carbonyl of amides are generally unreactive, so they react with only the most reactive reagents. That is why reduction of amides to the amines require LiAlH4 and does not work with H2, Pd/C or NaBH4.
Q: In chapter 22, there are several problems that ask us to nitrate a benzene ring. Sometimes the desired nitrated product has a nitro group on both the ortho and para positions, while sometimes, the nitro group just goes on either the ortho or the para position. If you want to put 2 nitro groups on the ring, do we use the same reagents (H2SO4 and HNO3) as if we just wanted to put 1 nitro group on? What if we wanted to put 3 nitro groups on (both ortho positions and the para positions)? The book seems to use the same procedure no matter what the desired product is. This doesn't seem very realistic. Or, in practical terms, does nitration just lead to a broad range of products that need to be isolated individually.
A: Yes, the same reagent is used for multiple nitration reactions. Nitration of a mono-substituted aromatic ring in which the substituent is an ortho/para director will give a mixture of ortho and para products; often the para product is slightly favored. This will be the case for almost any electrophilic aromatic substitution reaction. In the case of nitration, the nitro group is deactivating, so subsequent electrophilic aromatic substitutions will be slower than the first. The rate of the second nitration is usually slow enough that the reaction can be stopped at the mono-nitro stage; however, if the reactions time is extended, then bis-nitration occurs. So this is largely a matter of reaction times. The bis-nitro product will be further deactivated, so nitrating a third time will be even slower.
Chapter 21
Q: In Chapter 21, problem 21.30, there is 2-methyl-propanoic acid as the starting material. It first reacts with NaH, THF. Then LDA, then (CH3)3CCl, then H3O+. Does NaH would reduce the carboxylic acid to an alcohol? Then what would LDA do? The answer is 2 products: (CH3)2C=CH2 + (CH3)2CHCOOH
I am very confused about how the carboxylic acid was re-formed, and basically about the pathway to both products, could you please explain?
A: NaH is not a reducing agent but a base. It deprotonates the carboxylic acid to the carboxlate anion. LDA then does an alpha-deprotonation to give the enolate of the carboxylate- overall a di-anion is formed. One would expect this to undergoe alpha-substitution with a alkyl halide; however, the alkyl halide in this case is tertiary. So the enolate acts as a base rather than a nucleophile and affects an E2 elimination reaction, giving the alkene, (CH3)2C=CH2. When acid is formed the carboxylate is protonated to give the carboxylic acid back. It was a bit of a trick question.
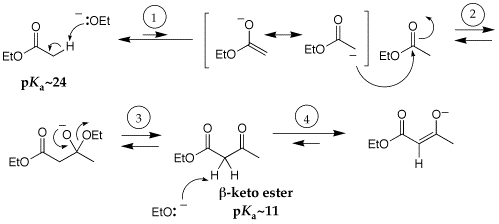
Q: I do not understand the last two steps for the mechanism of the Claisen Condensation, the deprotonation of the beta-keto ester and then the acidification. I know that it is related to the fact that the reaction is base promoted and not base catalyzed, but I don't understand why or how.
A: Below is the mechanism of the Claisen condensation. In step one, the base (ethoxide ion) deprotonates the ester to give an ester enolate. This equilibrium is not favorable because the pKa of an ester is ~24, while that of ethanol is ~16 (the conjugate acid of the ethoxide). The very small concentration of the enolate reacts with a second mol of ester in a nucleophilic acyl substitution reaction (steps 2 and 3) to give the beta-keto ester product; the formation of product regenerates ethoxide ion. However, the pKa of the product beta-keto ester is 11, which is much more acidic than the starting ester. So the regenerated ethoxide will deprotonate the product beta-keto ester much more favorablly than deprotonation of the starting ester. Since the product pKa is lower than that of ethanol, the equilibrium for deprotation of the beta-keto ester is favorable. Deprotonation of the beta-keto ester consumes the ethoxide, which is why the reaction is not base-catalyzed. A Claisen reaction must then be acidified to neutralize the beta-keto ester anion (to get back to the product of step 3). Think about the acidification step in the same way that a Grignard reaction must be acidified at the end of the reaction (I've called this the workup); when a Grignard reagent adds to a ketone or aldehyde, the initial product is an alkoxide anion that must be protonated (acidified) to the produce alcohol.
Chapter 20
Q: If an acid anhydride with different acyl groups were reacted with alcohol, what would be the ester that is formed? For example, the anhdride of benzoic and pentanoic acids reacts with methanol, would the methyl benzoate or methyl pentanoate be the product?
A: The reaction of an alcohol with a mixed anhydride would normally give two different esters. As such, the use of mixed anhydrides for the preparation of esters and amides is not very useful.
Q: What is the difference was between an acid catalyzed and an acid (or base) promoted reaction?
A: A catalyst increases the rate of a reaction but is not consumed in the reaction. So in the mechanism, the acid (or base) catalyst is regenerated so that it can continue to catalyzed the reaction. In the case of an acid or base promoted reaction, the acid or base is necessary for the reaction, but it is consumed. So it acts as a stoicheometric reagent rather than a catalyst.
Chapter 19
Q: How do you know when to eliminate and -OH group with acid/water/heat or with base, i.e. in problem 19.20 (h) base was used but in 19.24 (a) elimination was done with acid.
A: It usually depends on what (the leaving group) is being eliminated. Halides are usually eliminated by an E2 mechanism involving a strong base. Alcohols eliminate by an E1 mechanism in which the carbocation is generated with acid. This is preferred over E2 elimination because hydroxide is not a very good leaving group. An exception to the later is during the aldol condensation. Here the alcohol, which is beta to a carbonyl group, is eliminated with base because of the increased acidity of the alpha protons.
Q: How do you know that the hydroxyacid spontaneously cyclizes to lactone, as in problem 19.30? -- What makes it spontaneous?
A: Hydroxy acid that can lactonize to form 5 and 6-membered rings usually so so spontaneously. The ring size makes them spontanelous; 5 and 6-membered ring formation is usually a favored reaction.
Chapter 18
Q: In Chapter 18 #32, they outlined a synthesis in the solutions manual in which the last step was catalytic hydrogenation to reduce the alkene. In chapter 15, we learned that catalytic hydrogenation also reduces ketones and aldehydes to alcohols, but the book showed that hydrogenation did not affect the carbonyl group.
A: Normally the reduction of ketones and aldehydes by catalytic hydrogenation is slow compared to the reduction of alkenes. It often requires highly active catalysts and high pressures of H2. So alkene hydrogenation is much, much faster and the reaction can be stopped before carbonyl reduction. One exception to this is when the carbonyl is conjugated to an aromatic ring, but I am not sure you book described that.
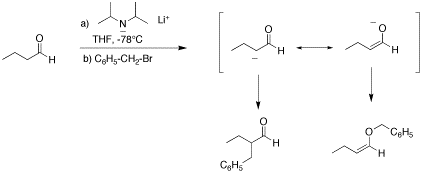
Q: This is in regard to the third part of question 1 on the second in-class quiz. LDA is a strong enough base to abstract an alpha proton and form the enolate (almost instantaneously)...page 762...in this case, if you look at the structure of the enolate formed, there are two nucleophilic sites (the alkene and the O minus ion). Given that part B has a primary alkyl halide leaving group, couldn't the strong nucleophilic character of the oxygen ion (essentially an alkoxide) do backside attack on the primary halide to form an ester?
A: Yes, there are two nucleophilic sites of an enolate, the alpha carbon and the oxygen. However, nucleophilic attack of the oxygen does not lead to an ester- it affords an ether. See below. We (nor the text) discussed O-alkylation but it is a frequest side reaction of enolate alkylation.
Q: Regarding section 18.12 on conjugate addition to unsaturated carbonyl compounds, why does the 1,2 addition have to be reversible for thermodynamic control? In 1,4 addition, should something be added 1,2 to see if it is reversible?
A: In the case of alpha,beta-unsaturated carbonyl, there is competing reactivity: 1,2- addition directly to the carbonyl carbon and 1,4-addition (conjugate) to the beta-carbon. 1,2-addition is generally faster than 1,4-addition. The 1,2-addition product is formed because kinetically it out competes the 1,4-addition product. With this in mind, the only way the thermodynamaically favored 1,4-addition product is observed is if the inital 1,2-addition reaction is reversible.
Chapter 17
Q: Should we know the reverse mechanisms?
A: Yes. The forward and reverse mechanisms are the same
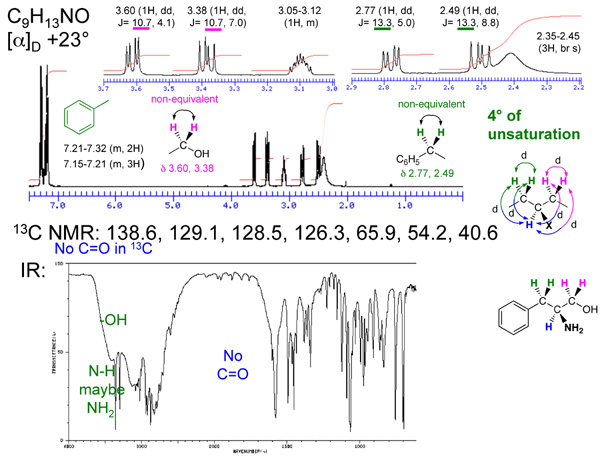
Q: Spectra problem of slide 124
A: Top; formula, C9H10O2 = 5 units of unsturation
IR: 1695, indicative of a carbonyl, 13C resonance at 191 indicates it is a ketone or aldehyde and not a carboxylic acid acid or ester; IR stretch at 1695 cm(-1) indicates that it may be a conjugated ketone or aldehyde.
1H NMR, two sets of doublets in the aromatic region that integrate to 2H's each is indicative of a para-disubstituted aromatic ring. Singlet at 9.8 ppm isan aldehyde, with no alpha-protons (no coupling constant). Quartet at 4.1 ppm (2Hs) is coupled to the triplet at 1.4 ppm (3Hs), which is consistent with a -CH2CH3. The CH2 resonance is shifted downfield consistent with an ether (-OCH2CH3).
Structure: p-ethoxybenzaldehyde
Bottom: formula: C10H12O = 5 units of unsaturation
IR: 1710 cm(-1) is a carbonyl. 13C NMR indicates that it is a ketone or aldehyde (207 ppm). No aldehyde peak in the 1H NMR; therefore a ketone.
1H NMR: Multiplet at 7.3 integrating for 5Hs is a mono-substituted aromatic ring. Quartet at 2.6 ppm (2Hs) is coupled to the triplet at 1.1 ppm (3Hs), which is consistant with a -CH2CH3. The CH2 resonance is shifted downfield consistant with it being next to an aromatic ring or a carbonyl group. Singlet at 3.6 ppm (2Hs) is consistent with a CH2 that is between carbonyl and an aromatic ring.
Structure is consistent with 1-phenyl-2-butanone
![]()
Q: Spectra problem of slide 125
A: Formula: C9H10O = 5 units of unsaturation
1H NMR resoanance at 9.8 ppm and 13C NMR resonance at 201 ppm indicate an aldehyde. The aldehyde resonance in the 1H NMR is split into a triplet by the CH2 at 2.7 ppm: -CH2-CHO
Multiplets at 7.3 and 7.2 ppm that collectively ingetrate for 5Hs are consistent with a mono-substituted aromatic ring. Triplets that split each other at 2.9 and 2.7 ppm are a -CH2-CH2-. They are shifted downfield, constant with them each CH2 being next to an aromatic ring or carbonyl. The triplet at 2.7 ppm is further split by the aldehyde proton into a doublet.
Structure: 3-phenylpropanal
![]()
Chapter 13
Q: I am a little confused about the complex splitting for 1H NMR. Problem 30(d) seems to have you adding the number of adjacent protons, where as in your example in the slides you had as multiplying the number of adjacentprotons. For example, if there were two non-equivalent adjacent Hs it would be a doublet of doublets or a quartet, is this not correct? The book implies that you would only have a triplet.
A: This has to do with complex coupling. If non-equivalent protons split a common proton, they do so independently. So as you say, one would expect a multiplet of multiplet. In the example you note, 13-30d, the proton that you are concerned about are those labeled b (see below), which have a chemical shift of 2.0, integrate for 2H, and are described as a pentet. A pentet implies 4 equivalent protons on the adjacent carbon, which is clearly not case. A doublet of quartets would be expected.
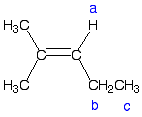
Complex splitting is observed when the two coupling constants are not the same. A doublet of quartets would be observed if J(ab) and J(bc) are not the same. However, if these coupling constants are the same, J(ab) = J(bc), then the pattern looks as if all the adjacent protons are equivalent. See the diagram below. On the right we have complex splitting. The two coupling constants are not the same and the pattern is a doublet of quartets. On the left, the two coupling constants are the same, so parts of the patterns overlap and what is observed is a simple splitting pattern.
.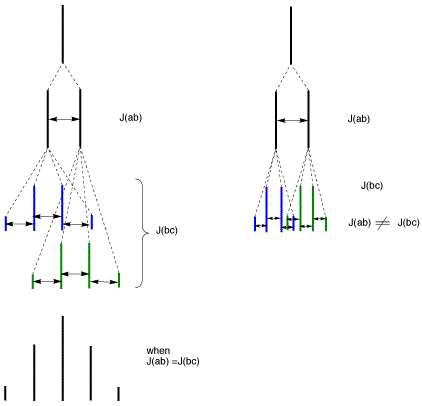
Also, a doublet of doublets in not the same as a quartet. A quartet (q) is a four line pattern with the spacing between the signals (the coupling constants, J) all the same and a 1:3:3:1 relative ratio. A doublet of doublets (dd), is a four line pattern with two different coupling constants. So the spacing two different spacings between the signals and the relative intensities is roughly 1:1:1:1.