Q: Will the Final exam emphasize the chapters 26-28 or will it also be more of a comprehensive final?
A: It will be comprehensive. My intent is that all the chapters will be equally represented on the final. There are overlapping themes in many of the chapters, i.e., Chapters 15 - 16 and part of 24 (electrophilic substitution), and chapters 19-23 (carbonyl chemistry). Themes somtimes reappear in subsequent chapters with expanded scope or emphasis (amine synthesis and amino acid synthesis). So the themes may be represented more so than the individual chapters.
Q: Will you be offering the alternate final exam on Wednesday, May, 3rd?
A: As stated on the syllabus, there will be no alternative final exam offered.
Q: Just to clarify, we do not need to know the amino chart in the beginning of chapter 26, right?
A: You do not need to know the structures amino acids per se (by popular demand), but you should be able to identify a hydrophobic, hydrophilic, basic and acidic amino acid when you see one.
Q: I was also unsure as to whether or not specific structure knowledge for portions of chapter 27 (i.e., phospholipids, prostaglandin, etc) would fall under the same category as the amino acid table.
A: As with the amino acids, you do not need to know specific structures, but you should be able to identify specific classes of compounds.
Q: On slide 369 we have a heading for section 26.9 with the (please read) caption, which we really know is secret code for "you will not be tested on this." Slide 370-377 then seem to fit under this heading. Are we to assume the material from slide 370-377 really do fall under the same caption as 26.9?
A: la·gniappe, [lan' yap] n, something given or obtained gratuitously or by way of bonus or good measure; a small gift given to a customer.
Chapter 28
04-27-2006
Q: I believe that the solutions manual made a mistake in synthesizing 5'-CTAG- 3' (Problem 28.49). I believe they actually made 3'-CTAG-5'.
A: You are correct. They synthesized the the DNA backwards. Be sure to remember that the chemical synthesis of DNA and peptides is done in the opposite deirection as the convention for writing a sequence.
Q: Why is chemical DNA synthesis done 3' to 5'?
A: The primary 5'-hydroxy group is less hindered and therefore more nucleophilic than the secondary 3'-hydroxy group.
Q: On slide 443: why does DMT-Cl react w/ the 5'-OH and not the 3'-OH?
A: For the same reasons as above. The DMT group is pretty big and reacts much faster with less hindered primary alcohols (5'-OH) than more hindered secondary alcohols (and not at all with tertiary alcohols)
Q: How can diisopropylamide act as a leaving group when it is the conjugate base of an extraordinarily weak acid? (p. 1084, step 3 of DNA synthesis)
A: The pKa of tetrazole is about 4.8. Tetrazole is an activator in this reaction and protonates the nitrogen of the phosphoramidite. This makes the diisopropyl ammonium a much better leaving group.
Chapter 26
04-24-2006
Q: Why do we use acetamidomalonate (with the amide group) rather than aminomalonate (with an amine)? The amide is ultimately hydrolyzed to the amine, but why begin with the amide?
A: A free amino group is nucleophilic and would compete in the SN2 reaction with alkyl halides.
Q: Why use the cyclic acetal protecting group during enantioselective catalytic hydrogenation (slide 359)?
A: It simultaneously protects both hydroxyl groups as acetal of formaldehyde. Protecting 1,2-diols as acetals or ketals is standard.
Q: Could you provide the mechanisms for the penultimate and ultimate steps of the derivatization with ninhydrin (slide 364)?
A: The penultimate step is the hydrolysis of an imine. So it would be the reverse mechanism as the formation of an imine (Chapter 19.9). The ultimate step is the formation of an imine with a second equivalent of nunhydrin.
Q: What is the purpose of amino acid analysis (determining the percent composition of each amino acid) if peptide sequencing (via Edman method) provides this information in addition to the linear amino acid sequence?
A: Sequencing is done on small fragments. The amino acid analysis is can be done on the entire protein. Since these are large macromolecules, determining structure is much more challenging and the error associated with each method is larger. Amino acid analysis is another piece of data that serves as a check for other methods. In the end, all the data should be reasonably consistent.
A: Could you provide the mechanism for the acid-catalyzed rearrangement of the anilinothiazolinone (ATZ) derivative to an N-phenylthiohydantoin (PTH)?
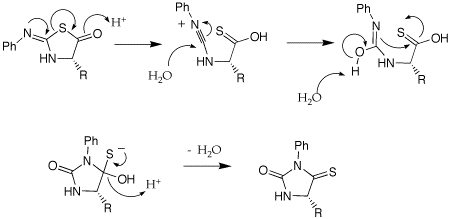
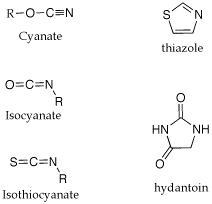
Q: Could you please explain some of the nomenclature used in the Edman degradation? (isothiocyanate, anilinothiazolinone, thiohydantoin).
A: I am not sure where the names come from. The attached gif shouws the structures of the parent ring structures. A cyanate is a R-O-CN; a isocyanate is isomeric in the placement of the R group, O=C=NR. A isothiocyanate substitutes sulfur for O, S=C=NR. The thiazolinone is based on a thioazole ring structure, the -one implies a ketone carbonyl within the ring and the anilino implies a aniline imine as part of the ring structure
Q: I do not understand why the Edman degradation has a length limitation. The book indicates that it is due to the build-up of unwanted byproducts, which I assume refers to the PTH-derivatives (?). If these are eluted from a column, however, does this not remove them from the system, allowing one to continue? What exactly limits this process?
A: The use of the an LC column is for analysis of the reaction not for purification of the product. The peptide and reagents are in a reaction vessel and the PITC is added and the pH adjusted to achieve the N-terminal amino acid cleavage and conversion to the thiohydantoin derivative. An aliquot is removed for the LC analysis and identification of the amino acid derived thiohydantoin. More PITC is then added to the reaction and the pH is cycled to get cleavage of the next amino acid (from the N-terminal end); however, the thiohydantoin from the previous cycle(s) is still part of the reaction mixture. After 20 cycles or so, the analysis become complex.
Q: What is meant by the "orthogonal protecting group strategy" (slide 378).
A: When there is more than one protecting group in a substrate, each protecting can be removed selectively without removing the others. In the case of peptide synthesis it is critical to be able to deprotect the alpha-amino group, without removing the protecting group on the carboxylate.
Q: Why is trifluoroacetic acid used for removal of the BOC protecting group? Could one instead use a mineral acid?
A: Trifluoroacetic acid (TFA) is used neat. Since trace water interfers with the peptide coupling step, it is desirable to avoid water during the peptide synthesis. TFA is actually low boiling and it is easy to get rid of.
Q: On page 1007 (peptide synthesis), why are steps 4 and 5 not combined into one? Both groups could be removed with acid (we learned in ch. 21 that ester groups are hydrolyzed by acid)
A: In principle it can be done in one step; however, aqueous acid is not as effective for removing a BOC group as neat TFA. The removal of the BOC group is actually not a hydrolysis. Q:
Q: Why are fibrous proteins insoluble in water? Do they not have hydrophilic amino acids distributed on their exterior?
A: The structure of fibrous proteins does not optimize the packing of the hydrophobic amino acids into the middle of the structure. I think it has more to do with the hydrophobic amino acids being exposed to the solvent. They also tend to be high molecular weight oligomers.
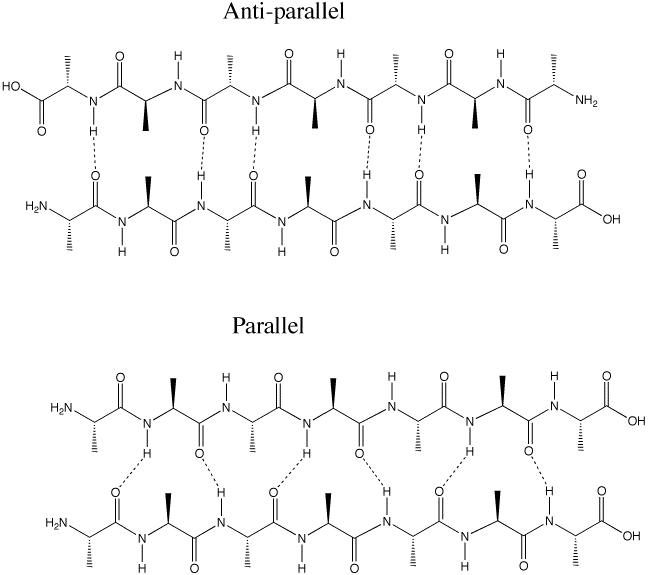
Q :Why is the antiparallel arrangement of the beta-sheet more energetically favorable than the parallel arrangement (p. 1011)
A: The hydrogen bonding between in the anti-parallel beta-sheet is better aligned than in the parallel. Hydrogen bonds are most stable with the O-H-N angle is 180 degrees. This can be achieved in the anti-parallel, but not in the parallel arrangement
04-10-2006
Q: On slide 352 where the basic and acidic amino acids are shown, why would acidic amino acids have more COO- than NH3+ and vice-versa for basic when the COO- is supposed to be more basic and the NH3 more acidic?
A: Both CO2H's of Asp and Glu are more acidic that the NH3+; so the CO2-'s would be weaker bases that -NH2. However, the overall charge state will depend on the pH of the solutions (see slide 354 on pI's; I should have drawn a arrow from right to left for increasing pH). If you are asking why I drew them in the completely ionized forms, it is probably because in peptides and proteins (which is really the more interesting situation), the a-amino carboxylate portions would be involved in amide linkages and are not in the zwitterionic forms. The side chains are largely in the ionic forms at neutral pH.
Exam 3 (04-07-2006)
Q: Will you be providing a "reagents bank" like you did last time? I'm finding that I can easily recognize what each reagent does when I see it, but being able to call up all of the reagents is a bit more challenging.
A: Haven't decided yet, but it would be safer if you are prepared for a challenge.
Chapter 25
04-28-2006
Q: On p. 948, it appears that the text relates R to D and S to L. Is this correct? I thought there was no relationship between R/S and D/L.
A: It is probably generally true for carbohydrates by virtue of the structural similarities between carbohydrates. However, R/S is a nomenclature system with rules. D/L has a physical meaning in the Fischer projection context. Cysteine is a good example where this breaks down. All of the amino acids are of the L-configuration which pertains to the position the alpha-amino group on the Fischer projection. Cysteine is R, while all the other amino acids are S.
04-06-2006
Q: for Chapter 25, do you want us to memorize the names of all the pentoses and hexoses from section 25.4?
A: I think you should be familiar with glyceraldehyde because all stereochemistry is related to it and it is of historical significance. You do not need to know the names or structures of any other carbohydrates, but you should be able to idenified their classification, i.e., a ketose versus aldose, furnaose versus pyranose, monosaccharide versus disaccharide, reducing versus non-resucing, etc). It would be helpful if you know that -ose means a carbonhydrate.
Q: I'm at the Fischer Proof section now and was wondering what you want us to note from this? I understand how he was able to go through deductive reasoning to figure out structures based off of optical activity, so was this section pretty much just to illustrate how incredibly much reasoning power went into early/current organic chemistry?
A: It was an illustartion of the use of the chain-lengthing reaction (the chain shortening could also have been used) in combination with the use of stereochemistry and stereochemical relationships and optical activity for structure proof.
Q: Why aren't polysaccharides considered reducing sugars if they have one free anomeric carbon at the end?
A: In principle, a polysaccharide should be classified as a reducing sugar if it is derived from an aldose and has a free anomeric end. Polysaccharides are very large polymers having MW on the order of 500,000 and 3,000 carbohydrate units per molecule of polymer. The reducing end of the polysaccharide is a small fraction of the carbohydrate present. So a 3000 carbohydrate polymer has a single carbohydrate unit which is a reducing end. It is likely that one can't get enough polysaccharide in solution on a molar basis to get an appreciable Tollins test that is visible.
Q: What is the use of dtermining whether a carbohydrate is a reducing sugar?
A: A reducing sugar gives a positive Tollins test. A carbohydrate gives a positive Tollins test if it can have an ring open form that is an aldehyde.
Q: Can you control whether you get alpha or beta product in disaccharide formation?
A: Only through the Knonigs-Knorr reaction (as well as variants to this reactions) can they be synthesized and the stereochemistry controlled for disacchardides. Nature synthesizes disaccharides and larger carbohydrates and controlls the stereochemistry enzymatically.
For monosaccharides the alpha and beta hydroxyl groups are in equilibrium, so there is no way to control the stereochemistry there.
Q: Is the Koenigs-Knorr reaction different from glycoside formation in any other way besides that it gives only beta product? Also, are there any limitations on the alcohols used in these reactions?
A: What is presented in Chapter 25 on the Koenigs-Knorr reaction is very fairly minimal. On can not make couple two carbohydrates together with acid to get a disaccharide and has to use the Koenigs-Knorr approach. There are restrictions to this reactions but most are specific to the particular reaction that is being done and the specific carbohydrates involved.
Q: Can the Koenigs-Knorr reaction be used for any carbohydrate?
A: In principle it can be used with any carbohydrate. If a 2-deoxysugar is used, the stereochemical preference for the glycoside formation is often lost.
Q: Slide 343: Is the reason that there is no open form of sucrose that both linked carbohydrates are glycosides? If so, how do they link so that the two anomeric carbons are connected? Also, why is the anomeric carbon an fructose number two instead of number one?
A: Yes, when the anomeric position is involved in a glycoside linkage, then it will not have an ring open form.
We really didn't talk about disaccharide synthesis much. While the anomeric hydroxyl group of the cyclic form has special reactivity, any hydroxyl group of a carbohydrate can be involved in a glycoside linkage.
The anomeric carbon is the one that is a carbonyl in the ring open form. For fructose, which is a ketose, that is carbon 2.
Q: Slide 331: You said in lecture that acetyl chloride could also be use in ester formation, would the product be the same but with chloride in place of the methyl group? When would you want to use one over the other?
A: No, you use acetyl chloride in place of acetic anhydride. They have equivalent reactivity and give the same product.
Chapter 24
04-28-2006
Q: For the Hofmann elimination, is Ag2O/heat OR a strong base like (CH3)3CO-K+ enough to eliminate the amine group?
A: I skipped the stuff about exchanging the counter ion with Ag2O. Treating the trimethylammonium salt with a strong base, such a t-butoxide, will give the Hofmann elimination
04-06-2006
Q: I was wondering if sections of Chapter 21 that we didn't get to cover for the last test will be on the upcoming exam (if so, which sections are they again?).
A: The material that we didin't get to in Chapter 21 was on the Chemistry of Amides. Some of that has resurfaced in Chapter 24
Chapter 23
04-06-2006
Q: Regarding slide 263, is the rxn with (H3C)3CO-K+ just a way to remove HBr to make the double bond?
A: Yes, Potassium tert-butoxide is my official E2 elimination base.
Q: In the reaction which gives alpha-beta unsaturated esters, acids and amines, where does the group Y come from and what is it representing? In the same reaction series, is R any ester or any amide? Also, how is R removed and why add it if only to remove it later? Are R and Y originally in the same compund?
A: The first part of the sequence is the HVZ top initally get the a-bromo acid bromide. We saw in Chapter 22.4 (ssee slide 234) that the a-bromo acid bromide can be converted into the a-bromo carboxylic acid, a-bromo-amide or a-bromor ester depedning on how you quench the HVZ reraction. Y represents these various permutations of the HVZ reaction, so Y is weither an -OH, NR2 (generic amide) or OR (generic ester). For the reaction immediately above shows the bromination of a carbonyl group (either an aldehyde or ketone) folloed by elimination to the a,b-unstaurated carbonyl. We only considered reaction at one side of the carbonyl and the R is just a generic organic group (or an H if it is an aldehyde)
Q: In the Claison Condensation, will you please explain how the deprotenation drives the reaction forward?
A: The Claisen reaction is reversible. If you take the b-keto ester product and add ethoxide to the keto carbonyl, you can break the C-C bond to give the ester and the enolate. When the product is deprotonated, the reverse reaction is prevented because ethoxide can not longer add to the negatively charge product. Deprotonation therefore stabilizes the product. Whenever you stabilize one side of an equilibrium, you drive the equilibrium reaction in that direction
04-05-2006
Q: During the aldol reaction, does water leave spontaneously, or is there a specific catalyst/reactant used to remove water and create a C=C double bond?
A: The dehydration occurs with either catalytic acid or base. The dehydration of of the beta-hydroxy-carbonyl product can occur during the aldol reaction if the reaction is done at room temp or above. At lower temperatures, the beta-hydroxy-carbonyl is stable.
Q: On page 875 of the text, the authors state that the first step in the reaction process is enamine formation from a ketone. Can the reacting enamine not be formed from an aldehyde?
A: They can be used with aldehydes but are more commonly used with ketones.
Q: When aqueous acid is added in the final step of the Stork reaction, will esters and nitriles present in the reacting species be hydrolyzed along with the enamine? If not, why? [this pertains to question 23.19 (a) and (c), page 875- the solutions manual indicates that these functional groups are not hydrolyzed]
A: The hydrolysis of nitriles and esters usually requires more vigorous conditions than for the hydrolysis of an enamine or imine back to the ketone (more conc. acid and usually heat). So these functional groups are safe during that step.
04-01-2006
Q: On slide 269 (23.5 Using Aldol Reactions in Synthesis), why is it that you get the b-hydroxy carbonyl or the alpha/beta unsaturated carbonyl from the aldol reaction. Is this the mixture of products that can result (it appears that the unsat. carbonyl is the dehydrated product of the b-hydroxy carbonyl??)?
A: We are not talking about getting a mixture of products although that is certainly a possibility. The aldol reaction can give two possible products. It can be stop at either the beta-hydroxy carbonyl stage. The dehydration can be catalyzed by either acid or base. The aldol reaction is often run under catalytic base conditions. If the aldol is done at lower temperatures, say 0° C or below, then the b-hydroxy carbonyl is the product. If the aldol reaction is run at room temperature or above, then the dehydration occurs top give the a,b- unsaturated carbonyl
Q: When we talk about enolates, are we only talking about enolate ions?
A: Yes, the word enolate refers to the enolate anion.
Q: LDA in THF at -78 degrees helps to cause E2 elimination as a strong base; but it also is used in the alkylation of ketones/esters/nitriles?? I am trying to understand slide 274 where is shows discrete (in situ) generation of an ester enolate with LDA; what purpose is the enolate serving in this purpose? Is it E2 elimination again or alkylation as well?
A: LDA can be used to affect an E2 elimination reaction. However, the way we have used LDA is to perform a-deprotonation of carbonyl compounds. On slide 274, once the enolate is formed it is undergoing a Claisen condensation with various esters. There is no E2 elimination on this slide.
Q: So, does the aldol reaction require that one of the reactants has alpha hydrogens whereas the Claisen condensation can do a reaction between a ketone enolate with an ester to form a 1,3 diketone?
A: The aldol and Claisen reactions are very closely related. Any of the enolates that we have discussed can be used for both of these reactions. The only difference is that the electrophile. For the aldol reaction, the enolate reacts with an aldehyde or ketone to give a b-hydroxy carbonyl (which can dehydrate to the a,b-unsaturated carbonyl). For the Claisen reaction, the enolate reacts with a ester to give a 1,3-dicarbonyl (rather that a 3-hydroxyl carbonyl which is the same as a b-hydroxy carbonyl). There is a parallel to the reactivity. The electrophile for the aldol is a ketone or aldehyde and the product is a b-hydroxy carbonyl. The electrophile for the Claisen is a ester (which is in the next higher oxidation than a ketone or aldehyde , the electrophile for the aldol) and the product after reaction with the enolate is a b-keto carbonyl, which is in the next higher oxidation state than a b-hydroxy carbonyl from the aldol reaction.
Q: On slide 274, how do we know that the Claisen condensation always breaks the bond between the C and the OEt?
A: The Claisen condensation is a nucleophilic acyl substitution reaction between an enolate and an ester. When the enolate adds to the ester, a tetrahedral intermediate is formed. The carbonyl is the re-formed when the EtO(-) is ejected as a leaving group.
Q: On slide 277, what is the purpose of having EtONa and EtOH in the second step when the bromoalkene is added? (what is that reaction?)
A: This is a a-substitution reaction. The substitute reaction of the hydrogen for an alkyl group must go through the enolate. The EtONa is to generate an enolate.
Q: If decarboxylation is a loss of CO2 why does the whole of the CO2Et get removed in the addition of H30+ and heat in the reaction on slide 277?
A: This is a two stage reaction. The first is the acid catalyzed hydrolysis of the ester to the carboxylic acid. The second is the loss of CO2; overall, the CO2Et is lost as CO2 and EtOH
Q: When you were drawing the reaction for the Robinson Annulation you mentioned that of the four alpha carbons/hydrogens one is far more acidic and it was the one between the two carbonyl groups - why was this not the one extracted by EtO-?
A: You are talking about the intermediate after the Michael addition that undergoes intramolecular aldol to give the cyclohexenone product. There are at least four different enolates that are possible from that intermediate. The major enolate from deprotonation between the ketone and ester can react with the other ketone, but this would give a disfavored four member ring aldol product. Deprotonation at either of the methyl groups next to the ketones, gives an enolate that can react with the other ketone to give a much more favored six-membered ring aldol product (see next question). Even though these enolate are present in much lower concentrations, they react to give more stable products, so the aldol to give six-membered ring is more favored than to give the four-membered ring.
Q: Would two products not have been possible from the Robinson Annulation you showed us (for ex. two products with alkene double bond in two different places?)
A: For the Robinson annulation product on slide 283, it is possible for the double bond to be in conjugation with the ester. However, the base catalyzed dehydration of water that yields this double bond results from a-deprotonation and elimination of hydroxide. One would expect deprotonation alpha to the ketone rather than the ester because it is the more acidic position.
If you look at this intermediate on slide 282, the carbon fragments are color coded. What is shown is the blue methyl ketone undergoing aldol reaction with the green ketone and dehydration to give the product. It is possible that the green methyl ketone can undergo aldol reaction with the blue ketone followed by dehydration to give a cyclohexenone was not shown on the slide in the notees (see below). This is certainly a feasible reaction, which I did not consider. Thamks for pointing this out.
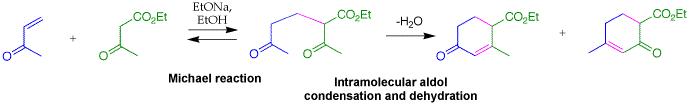
03-31-2006
Q: On page 863 (middle of the page), the reaction is a mixed aldol reaction; why does it produced an unsaturated product using the same reagents (NaOEt and ethanol) while the reaction at the end of the chapter on p. 881 (1c) shows the
product not having been dehydrated? (yet, the reactants are still the same)
A: As I noted above, the conditions of the aldol reaction determine if the product is a b-hydroxy carbonyl or the a,b-unsaturated carbonyl. You need to be able to recognize that two products are possible for the aldol reaction.
Q: Is the effect of acetoacetic ester synthesis the addition of three carbons to the carbon chain?
A: Acetoacetic ester is initally a four carbon unit (not including the OR of the ester). After decarboxylation, a three carbon unit is added.
Q: Can cycloalkanes be made with acetoacetic ester synthesis like that can intramolecularly with malonic ester synthesis?
A: Yes
Q: Is the enol-keto tautomerism essentially equivalent to the relationship between resonance structures?
A: No! Enol-keto tautomers there is a movement of a proton. You can only move electron when draeing resonances structure; atoms can not move
Q: In the carbonyl condensation reaction, why do we show the carbonyl as an enolate ion and not an enol?
A: This depends on the on the reaction conditions. If it acid catalyzed, then the enol reacts, but this is less common and less reactive. For the base catalyzed reaction with ketones or aldehydes, you can draw either the enol or enolate- the enolate is more reactive than the enol, so I usually show the enolate reacting. The pKa pf beta-dicarbonyl compounds is low enough that the enolate is the reactive species when treated with base. If you use a strong base such as LDA, the the carbonyl compound is completely deprotonated and the enolate is generated.
Q: Is it true that the starting carbonyl in a carbonyl condensation reaction is an electrophile that than becomes a nucleophile upon attacking the second carbonyl compound?
A: It becomes a nucleophile (and stops becoming a electrophile) upon enolate or enol formation.
Q: Why is the aldol reaction between two ketones the first reaction listed in Chapter 23 if it is so unfavorable? Is it still a reliable reaction?
A: I don't thing that the way it was presented specified a ketone per se. It could also be an aldehyde. The self-condensation of either a ketone or aldehyde is the basic reaction
Q: In the acid catalyzed dehydration of aldol, it is possible to go directly from the first resonance structure enol to the final product with E2 elimination and bypass two or three steps, right?
A: If you mean to bypass the resonance structure with the the negative charge on the alpha-carbon, yes. Remember, that neither resoancne structure is truly correct- the the actual structure is a hybrid of the resonance structures.
Q: On the last reaction of slide 261, should there be two equivalents of the reactant to do a carbonyl condensation reaction followed by an aldol reaction?
A: The slides numbering has gotten messed up so I am not exactly sure what reaction you are talking about. If it is the aldol reaction of cyclohexanone, then yes, there should be a two in front of the reactant.
Q: Is the carbonyl condensation reaction/mechanism discussed first in Chapter 23 an overall reaction/mechanism for carbonyl compounds or is it its own separate reaction? It was not listed in the summary of reactions at the end of Chapter 23, so I was just wondering what its status was.
A: The reaction shown on page 855, Fig 23.1 is the base-catalyzed aldol reaction. It is the general reaction that is listed at the aldol reaction in the Summary on page 881. It happens to be listed as three specific types.
Q: What were the conditions for that you gave in slides 262 and 263 (for ex. if one partner has no alpha H or if one of the carbonyl species is way more acid than the other)? Were they conditions for producing one product in the mixed aldol reactions as opposed to 4?
A: The mixed aldol reaction (slide 264 in the latest version) gives four possible products. This uses the base-catalyzed conditions shown. The way to get just one is to discretely generate the enolate from one of the aldehydes (lets say pentanal) with LDA in THF at -78° C; this completely convert pentanal into the enolate. After the enolate is formed, the other aldehyde is added (in this case propanal). This will give just one of the aldol products: 2-(1-hydroxyproyl)penanal (top right) because the pentanl was convereted to the enolate which is going to act as the nucleophile and propanal is the electrophile in the reaction.
Chapter 22
04-06-2006
Q: When, if ever, is the haloform reaction useful?
A: There are a few examples in the book when a methyl ketone is converted to a carboxylic acid (by loss of the methyl group through the iodoform reaction). Overall, this is not a useful reaction
Q: In the malonic ester and acetoacetic ester syntheses, the products are an alkylated carboxylic acid and an alkylated methyl ketone, repectively.
a. Is it correct that you first make an enolate ion with LDA (or another base) and then react it with an alkyl halide and then use acid to get the final product?
A: The text did not emphasis the formation of the enolate with LDA, although this is the most reliable way to form enolates. The alkylation step can be done with catalytic base. The decarboxylation of the alkylated product is done with acid treatment.
Q: b. Could you, and if so would you, ever stop after the reaction with the alkyl halide?
A: It depends on the goals of the synthesis. In principle, yes, if you want a substituted malonic ester. But in general, one of the ester groups is decarboxylated
Q: Can you react the starting material with acid without first reacting with the alkyl halide?
A: No. The point of the other ester group is to improve the reaction by increasing the acidity of the substrate. One can alkylate an ester if the enolate is generated with LDA
Q: Have we learned how to make a malonic ester or would it be given as starting material?
A: Yes we have learned who to make malonic esters (it is the product of a Claisen condensation of ethyl acetate and diethylformate) but it is usually given as a starting material
04-05-2006
Q: on slide 248, for the beta-keto carboxylic acid and decarboxylation, you used HCl and heat, but the book just says -CO2...are they the same?
A: This is a hydrolysis of the esters, so you need aqueous acid. So it should be H3O+ or HCl and H2O plus heat. The aqueosus acid hydrolyzes the ester to a carboxylic acid which then loses CO2.
03-20-2006
Q: I was reading section 23.4 on the dehydration of aldol products and I noticed a small discrepancy between your mechanism you did in class and the book's mechanism. In the book, it says that the carbonyl oxygen is protonated first to form the enolate ion, followed by the protonation of the beta -OH group. However, your notes show the beta -OH group protonation followed by the carbonyl protonation. If your mechanism is correct, why would the -OH group be protonated first? Wouldn't the carbonyl oxygen be more basic?
A: I apologize; I try to be as consistent with the book as possible.
I will first explain why in this case the order doesn't matter, as opposed to other mechanisms such as the acid-catalyzed hydrolysis of a ester where the order of events does matter. I will then explain why the book's mechanism is better.
In the case of the acid-catalyzed dehydration, the order of enol formation and hydroxyl group protonation doesn't matter much because these two events must both happen before dehydration can take place and one is not dependent on the other. Since both the hydroxyl protonation and enol formation are part of an equilibria, the desired hydroxyl protonated enol will be obtained some percentage of the time and this intermediate will undergo dehydration. This is opposed to other reactions such as the acid-catalyzed hydrolysis of a ester, which is also a muti-step mechanism that are each part of an equilibrium. In that case, protonation of the carbonyl group activates it towards nucleophilic addition; so the second equilibria (nucleophilic addition to the carbonyl) is dependent on the first step of the mechanism (protonation of the carbonyl group).
The mechanism in the text for the acid-catalyzed dehydration of an aldol is better than the one I drew on the board. The enol formation is acid catalyzed, so the acid is not consumed in this step. Protonation of the hydroxyl group consumes the acids until it is regenerated upon dehydration. If you protonate the hydroxyl group first (the acid is not regenerated until after dehydration), a second equivalent of acid is needed to catalyzed the enol formation. So the out of order mechanism that I drew on the board is actually sloppy in terms of keeping track of the catalyst.
Thank you for pointing that out.
03-19-2006
Q: For problem 22.27 (a), if the compound were instead meta-bromo-(3-oxobutyl)benzene, could one use an acetoacetic ester synthesis with meta-bromo-benzylbromide? (i.e., I am guessing that the aromatic bromine would not interfere with the reaction and that acetoacetic ester would react exclusively with the benzylic bromine)
A: Yes, that would be fine. The aryl bromide ewould be unreactive to the malonic acid synthesis
Q: Are there any limitations to the reagents that can be used in malonic/acetoacetic ester syntheses besides the general limitations of all SN2 reactions?
A: No, there are no general restrictions other than the limitations of the SN2 reaction
Q: Why are vinylic protons non-acidic even when the proton is on a carbon that is alpha to a carbonyl? Alkane protons which are on a carbon that is alpha to a carbonyl are acidic, and alkane protons are less acidic than alkene protons. (see problem 22.33 for clarification)
A: Normally a vinyl proton is more acidic that an alkyl proton; however, both are still very weak acids. The major effect for the increase in acidicity of protons on the alpha carbon to a carbonyl is that the anion can be stabilized through resonance into the carbonyl pi-bond. The alpha carbon of a ketone is sp3 hybridized but upon deprotonation it becomes sp2 hybridized; this puts the pair of electron in an unhybridized p-orbitals and allows the pair of electron (and charge) to be delocalized into the carbonyl pi-system. It is then analogous to the allyl anion from last semester but much more stable. If the alpha carbon is part of a vinyl group as in the example in problem 22.23, the geometry is not right for delocalization of the charge into the carbonyl group. In order to delocalize the charge into the carbonyl, the alpha carbon of 22.33 would have to become sp hybridized upon deprotonation. For what ever reason (I really don't know), there is a very high barrier to this. There is a much larger change in geomtry for the later.
03-15-2006
Q: For Meldrum's acid, is it the ring system that makes this compound more acid (ie. the negative charge can be distributed in the ring system as well as the carbonyl groups?)?
A: The ring is important and in a way helps to better distribute the negative charge, but it is not distributed throughout the entire ring. The ring holds both carbonyl groups co-planar which is the best conformation for delocalizing the charge into both carbonyl groups.
Q: In the HVZ reaction mechanism we did for slide 236, how does the first step
occur where PBr3 replaces OH?
A: Mechanism is very similar to the replacement of and alcohol with Br by PBr3 (see slide 98 from Ch. 17), rather than Br- participating in an SN2 reaction, and it in a nucleophilic acyl substitution reaction (similar to acid chloride formation with SOCl2). See below.

Q: And in the second step of the aforementioned reaction, where does the H come from that protonates the O on the carbonyl?
A: It is actually the proton of the carboxylic acid. Acid bromide formation product a phosphoric acid as the by-product of PBr3
Q: Does enol formation favor C=C at the less sterically hindered site (i.e., the left most compound on slide 233)?
A: No. Since this is an equilibrium, it is governed by the thermodynamics. The enol that will be favored is the one that is thermodyanmically the most stable, So the more substituted enol is favored because the C=C bond that is most stable is the more substituted.
Q: On slide 229, when you showed us the reaction of enol to alpha-substituted product, you said that the acid catalyzed reaction would come before - would it be fine for the base catalyzed reaction to occur in place of the acid catalyzed reaction?
A: Alpha-substitution of an enol can go by either acid or base catalysis
Q: It is possible to isolate the enol intermediate, right (even though the equilibrium lies on the side of the keto form)?
A: No, the concentration of the enol is low and the rate of the equilibrium is too fast for one component to be isolated. It is generally difficult to "isolate" one component of an equilibrium because it will interconvert to the others and the equilibrium mixture will eventually be restored. If the rate of the interconversion is slow, then is is possible to isolate one component, but it will eventually re-equilibrate.
03-13-2006
Q: Page 830 of the text mentions that "aldehydes, ketones, esters, acids, and amides can be converted into enolate ions by reaction with LDA." What about acid chlorides and anhydrides?
A: This is a good example of the basicity versus nucleophilicity. These parameters must also be measure in the contexts of the rest of the system. Acid chlorides and anhydride are generallt so reactive toward nucleophilic acyl substitution, that it would be very difficult to find a base that would rather act as a base and do an a-deprotonation over acting as a nucleophile and adding to the carbonyl. Although I can recall a very few exampled of enolate formation and anhydride, these are not compounds that enolates are generally formed from.
Q: Could you please explain the acidity order amides > nitriles > esters > ketones. Furthermore, why is the diester less acidic than the dinitrile (the mono-ester is more acidic than the mono-nitrile.
A: The amide is less acidic because the nitrogen resonance with the carbonyl reduces the ability of the carbonyl to stabilize the negative charge of the enolate. The same is true of the ester, although the resonance of esters is generally much weaker than for an amide. The acidity of nitriles also puzzles me and I always think that they should be more acidic than esters; I presume that the nitrigen is simpley not as good at stabilizing a negative charge as an oxygen.
I don't know for sure about the acidity of 1,3-dinitriles versus 1,3-diesters but I believe that it has to do with the conformation. In order to get the maximium effect from both carbonyl groups (or both nitriles) for lowering the pKa of the a-hydrogens, they have to be co-planar. What we observe for the pka is a weighted average of all conformations present. A percentage will have both groups co-planar and a precentage will not. Since the nitrile has such low steric demands, there will be a larger percentage (than for the diester) of the conformation where both nitrile groups are co-planar and able to cooperativly stabilize the negative change.
Q: Can carboxylic acids lose their alpha proton upon reaction with LDA? They will readily lose their OH proton, acquiring a negative charge. This negative charge will reduce the acidity of the alpha proton. I guess the proper wording of the question is: is the pKa of a deprotonated carboxylic acid less than 40 (the pKa of LDA)? If so, the carboxylic acid will be doubly deprotonated to its dianionic form.
A: Your reasoning is correct. Two equiv. of LDA will give the dianion of a carboxylic acid.
Q: Does the carbonyl bond energy account for the fact that electrophiles typically react with deprotonated carbonyls as alpha-keto carbanions rather than vinylic alkoxides? Is this the same reason why the keto form is favored over the enol form in their tautomeric equilibrium?
A: Not entirely. This is a very complex questions. As it turns out, the reactivity, C versus O can be influenced by the nature of the electrophile, the counter ion of the enolate and the solvent. The Hard-Soft Acid-Base principles are often used to account for this. The O is the hard site the C is the soft site. Li counterion promotes C reacitvity because Li+ is strongly asscoaited with the O- and thereby reduces O reactivity (there is significant covalent character to the Li-O bond). Less coordinating counter ions, such as K+ promote O reactivity. Diethylether does competes less well for coordination with the counter ion than THF, so it ether promotes more C reacitivity and THF promotes more O reactivity. DMSO gives even more O reactivity. The better the leaving group, the more O reactivity is observed. So alkyl chlorides give more C-reactivity, while tosylates give more O-reacitivity.
Q: Is the haloform reaction the exact same reaction in section 22.3 of the text (p. 824)? They simply waited to discuss polyhalogenation until section 22.7? Does haloformation only apply to methyl ketones? Why can fluoroform not be generated by this reaction? What is the usefulness of a haloform? I know that deuturated chloroform is used as a solvent in 13C NMR. Would this be a useful way to produce deuturated chloroform (if the reaction was done in D2O)? Also, I know that chloroform is useful for cyclopropanation (with regards to this, why does a deprotonated chloroform molecule eject chlorine to form a carbene? This seems like rather strange chemistry..). Are chloroforms (or the other haloforms) useful for any other reactions?
A: In a sense, it is the same. For section 22.3 it is a base catayzed halogenation of an enol. The haloform reaction involves a higher concentration of hydroxide and the hydroxide is consumed when CX3 is hydrolyzed. It is mechanistcally related, but with the higher hydroxide concentration, the haloform reaction may actually go through an enolate reather than an enol. The ejection of the CX3- is unique to methyl ketones, although the base promoted a-halogenation occurs with any ketone or aldehyde.
F2 is a fluorinating agent but it is an out-of-control one that actually needs special equipment. F2 will fluorinate just about anything and eventually replace virtually and C-H bond with a C-F. There are specialized fluorination reagents, but these do not fit well with the rest of the section.
I don't know how CDCl3 is made. The proton of chloroform is acidic, so I suspect they make CDCl3 from CHCl3 and base in D2O and exchange H for D.
The chloroform anion does decompose to dichlorocarbene, although I am not sure of the exact reactions conditions. It may be that dichlorocarbene formation is slow at room temperaturem however it is probably a side reaction. It is a strange reaction and is mechanistically similar to Cannizarro reaction. I can think of anything particulalrly usedul about haloforms other than as dihalocarbene sources.
Q: Why is it that factors that increase the carbonyl dipole moment increase its acidity and vice-versa for those which reduce the dipole moment? If the carbonyl dipole moment is increased, there is more positive charge on the carbon. Does this cause electron density on the alpha carbon to shift to the carbonyl carbon, thereby reducing the affinity of the alpha carbon for its protons?
A: Yes, that is the correct reasoning. The dipole pulls electron density from the alpha C-H bond and makes deprotonation easier. This is the inductive effect component. The resonance effect also plays a large role. Since an ester O or amide N is involved in resonance interactions with the carbonyl, the degree to which the enolate can be stabilized through resonance is decreased. This is probably the larger effect.
Q: Could you explain why benzylic and allylic groups are more reactive under SN2 conditions than alkyl groups? Also, why are vinylic groups unable to undergo SN2 reaction? The text says that backside approach is stereochemically prevented, but a model of a compound such as bromoethylene does not appear hindered.
A: We often draw the Walden inversion as a symmetrical transitions, that is the Nu-C bond is half formed while the C-X bond is half broken; this would mean that there is little if any charge build up on the carbon undergoing nucleophilic attack. This picture is not always the case. Very often C-X bond breaking is more advanced in the transition state than Nu-C bond formation and vice versa. This will leave a partial charge character on the carbon. As the C-X bond breaks in the transition state and the charge that is being generated on the carbon (either positive or negative) is stablized by the pi-bond of the vinyl or aryl group.
You could argue that backside attack of a vinyl halide is slightly more or slightly less hindered than for an alkyl halide. I don't think that sterics are the major issue. For the Walden inversion, the transition state geometry looks like a trignonal bipyramid geometry. This requires a "reorganization" of the moleculalr orbitals and this is part of the transition state energy. The analogous Walden inversion for a vinyl halide would require a square planar transition state geometry and my feeling is that the reoranization of the MO's would probably be significantly more costly energically.
Q: How does one control whether malonic ester is mono- or dialkylated?
A: With stoichiometry. The advantage is that the second alkylation will be slower than the first for steric reasons.
Q: The text mentions that decarboxylation can occur only for malonic acids and beta-keto acids. Can it also occur for other species with a carbonyl beta to the carboxylic acid such as beta-amido acids, beta-nitrile acids, and beta-ester acids? This nomenclature is probably incorrect. If it is unclear what I mean, I will attempt to rephrase the question.
A: Sure, anything that can stabilize the anion that is formed upon decarboxylation will work. The decarboxylation of a b-cyano acid is well known and in fact this is a variation of the malonic acid synthesis. Even a nitro group will work.
Q: Problem 22.12 on page 837 indicates that trialkylated carboxylic acids cannot be prepared. However, what if the product of the malonic ester synthesis (a dialkylated carboxylic acid) is treated with LDA to generate the enolate ion and is then reacted with an alkyl halide? Would this be an effective method of producing a trialkylated carboxylic acid?
A: Yes, that is a reasonable way to syntehszie such as a compound, although that is no longer a malonic acid synthesis.
Q: Could you explain why ethyl 3-oxobutanoate is also called acetoacetic ester? Which part of the molecule is the "aceto" part and which is the "acetic"? Also, is it necessary for the ester groups to be ethoxide in both the malonic ester and acetoacetic ester syntheses? (or can they be other groups)
A: This is another reaction that is probably over 100 years old and acetoacetic acid was probably named before there was systematic nomenclature- so the name is strange. This is named as if it a 2-acetyl substotuted acetic acid. See attached gif.
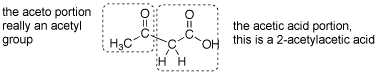
Q: On page 840, the text mentions that a nonprotic solvent must be used for direct alkylation of monocarbonyl compounds. Why is this? Also, how is what is discussed on page 840 and 841 (direct alkylation) different from what was discussed on pages 834-5 when the SN2 reaction of an enolate with an alkyl halide was mentioned?
A: If you use LDA to generate the enolate, you must use a non-protic solvent because a protic solvent will be deprotonated by LDA. So if you use LDA in ethanol, what you really have is lithium ethoxide, not LDA.
Direct alkylation as described at the end of the chapter is not conceptionally different from the base catalyzed enolate formation described at the beginning of section 22.8, except for the reaction conditions are considerable different. When the enolate is generated with NaOEt in EtOH, the concentration of the enolate is related to the relative pKa's of the base and carbonyl compound. So enolate concentration is fairly low. For direct alkylation with LDA, the enolate is generated descretely and quantitatively before the electrophile is introduced. These are much more reactive conditions. We will see the value of this more in the next Chapter.
Q: The text also mentions on page 840 that ketones, esters, and nitriles can all be directly alkylated. What about acid halides, acid anhydrides, and amides?
A: Acid chorides and anhydride can not be used to form an enolate. If the a-protons of an acid chloride is deprotonated, the Cl elimates: R2HCC(O)Cl -->R2C=C=O this is called a ketene which has unique chemistry of it own (See ketene gif). An anhydride would do the same chemistry. Enolates of amides are fine so long as there are no N-H bond invloving the amide nitrogen. So an enolate of a tertiary amide can be formed.
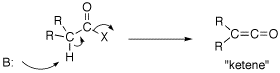
Q: The text mentions on page 841 that the major product of direct alkylation reactions generally occurs at the less-hindered (less-substituted) position. However, the enolate ion is the nucleophile and I thought that there were few restrictions on the nature of the nucleophile for SN2 reactions (the nature of the alkyl halide, by contrast, is of supreme importance). I feel that a better rationalization of the major/minor products in this instance is that the enolate ion formed at C2 is destabilized by the electron-donating methyl group relative to the enolate ion at C6. What do you think about this?
A: The text actually does not present this well. Your initial reasoning that thisis not about the sterics of the alkylation reaction is correct. It is all about the sterics of the deprotonation step. The deprotonation of an unsymmetrically substituted ketone with LDA in THF is under kinetic control since the deprotonation is essentially irreversible. So the less hindered a-proton is deprotonated fastest because it is more accessible to the base, leading to the less substituted enolate. See enolate gif. Deprotonation of the more hindered a-protons is slower but leads to a more substituted enolate which is thermodynamically favored. This is formed when the enolate is generated under reversible conditions, such as NaOEt in ethanol; this can be less selective because the ration of the more and less substitute enolate will be a function of their relative stabilisty. So this is the real value of generating enolates with LDA in THF at low temperature, you get only the kinetic enolate.
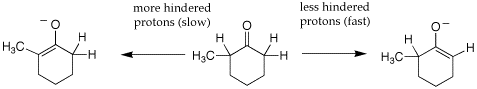
Q: The text mentions that alpha-halogenated products can be dehydrohalogenated to give alpha-beta unsaturated carbonyl compounds. Is this a good method of preparation of alpha-beta unsat. carbonyls? I thought that this reaction would give polyhalogenated products (if possible) and would therefore disallow dehydrohalogenation to form the alpha-beta carbonyl. Also, for ketones, since there are two sets of acidic hydrogens, one may have an even worse situation.
A: The polyhalogenation is more of a problem wih the base induced alpha-halogenation. These reactions are done under very basic conditions, so the enol formation is very rapid.
The reactions that we learn are fine on paper. Sometime there are disconnects in textbook; as I noted today the HVZ reaction is over 100 years old. There are more modern reagents and reaction conditions for many of these reactions that are vastly improved from the 1890's vintage and can control which side of the carbonyl is the site for substitution and eliminated problems of multiple reactions. The alpha-bromination followed by elimination to give a a,b-unsaturated ketone is fine paper chemistry, but it is no longer the first thing that comes to mind if one really wanted to do that reaction.
The descrete formation of an enolate eliminates the issue of multiple reactions because the carbonyl is treated with an equal molar equiv. of the base to generate the enolate. Once the enolate reacts with an electrophile, there is no longer any base around to form a new enolate. So the reaction is controlled by stoichiometry.
Q: Why is the HVZ reaction needed to alpha-brominate carboxylic acids? The text states that acids, esters, and amides don't enolize to a sufficient extent. (a) Why don't they? (b) I thought it didn't matter what minute amount of enol was present, simply that there iis some which can then react with an electrophile, thereby driving enolization forward. Also, can carboxylic acids be chlorinated and iodinated? Can carboxylic acid derivatives be halogenated? (it appears that only carboxylic acids can undergo the HVZ reaction)
A: This is likely to be a issue of rates. The amount of enol for a ketone or aldehyde is a small fraction of a percent. The pKa's of acids, esters , etc, are at least 7 pKa units less acidic. It is also likely that acid catalysts doesn't accelerated enol formation appreciably with acid derivatives to accelerate the rate of alpha halogenation enough to make the reaction useful.
I imagine that there is an anlogous set of reactions for a chlorination although I can not say that I actually remember seeing such a reaction. I do't noe of an anlogous reaction for a-iodination.
Amides would not work under the HVZ reaction conditions for sure since they are incomparible with Br2 and PBr3. PBr3 will dehydrate the amide to a nitrile and Br2 often reacts directly with the nitrogen.
I do not recall ever seeing the alpha bromination of esters and nitrile that did not involve enolate formation.
Q: Why would one need to isolate the enolate ion (by using LDA). Could one not simply use aqueous acid to catalyze enolization. An intermediate in base-catalyzed enolization is the enolate ion. Wouldn't this intermediate be able to undergo the desired reaction (e.g., alkylation)? What benefits are there from isolating the enolate?
A: Enolates are much more reactive than enols so they give higher yields and shorter reaction time. When one forms the enolate specifically, the problem of products derived from multiple alkylation is eliminated. We will see in the next chapter that forming descrete enolates is critical for what is know as a cross-aldol condensation. One does not isolate the enolate, it is generated discretely, then the electrophile is introduced to the reaction. This is referred to in situ.
Q: What did you mean by: "When one forms the enolate specifically, the problem of products derived from multiple alkylation is eliminated." How does an enol give multiple alkylation products whereas an enolate ion gives but one.
This is an issue of stoichiometry. Once the enolate is formed, the base is consumed. After the enolate reacts, subsequent enolate formation can not occur because there is no longer any base present. Under the catalytic conditions, the product can be enolized again and react with the electrophile again, although the rate of the second alkylation reaction is usually slower than the first.
Q: Someone brought up in class today the question of whether enolate ions could be formed using sodium hydride. The text says sodium hydride is capable of completely effecting enolization (p. 830). Also, you mentioned that butyl lithium and sodium amide can not be used for enolization because they are too nucleophilic. How can one assess a base's nucleophilicty. For example, how can one determine if a base is nucleophilic or non-nucleophilic. How can one determine whether a nucleophile is basic or non-basic. (as a side question, sodium amide, like LDA is also called an amide. I didn't know if you had figured out why. I have looked (unsuccessfully) on the internet for an answer to this curiosity)
A: I thought I agreed that sodium hydride could be used, although I wasn't sure of the pKa. It is certainly stronger than an alkoxide.
It is difficult to intuitively know what is going to be nucleophilic or basic. This is something that is learned through experience. As it turns out, many of bases such as hydroxide and amide are more basic than they are nucleophilic. However, they are nucleophilic enough to give significant side reactions. Amide would be unacceptable for formation of an ester enolate. Sodium amide can be used form ketone enolization, but it is inconvenient to prepare, relative to LDA.
I have also wondered about the nomenclature of amide but have never found a reason for it. It is unfortunate because it can cause confusion.
Exam 2 (03-01-2006)
02-27-2006
Q: I just have a quick question regarding mechanisms for tomorrow's exam. Should we know in detail all mechanisms or should we mainly focus on the ones you actually drew and discussed on the board during class? are those the more emphasized mechanisms in the chapter - should we study those in particular?
A: Many of the mechanism are very similar and there were some that I did in class because they might help you understand (and remember) the reactions. As for the last exam, the most likely mechanisms are those that exemplify a major theme of the material.
Chapter 21
02-27-2006
Q: Do we need to know reactions or mechanisms for acid anhydrides since we did not talk about them?
A: They are exactly the same as for acid chloride. just substitute a O2CR group for
the Cl.
Q: I was told of a web site that had unique/fun ways of remembering some of the mechanisms. I thought it was too great to not send to you, so if you get a chance check out the link below:
http://www.oir.ucf.edu/chemistry/s02/frameset41.html
A: Thank you . Unique is a good way to describe it. However, can you sing the song backwards as well?
Q: Does NaBH4 not reduce any of the carboxylic acid derivatives discussed in Chapter 21? And thus it can be used to reduce other ketones and such in the presence of these carboxylic acid derivatives?
A: It does not react with esters, amides, nitriles or carboxylic acids. Acid chlorides are incompatible with the NaBH4 reaction conditions which is done in ethanol and will give the ethyl ester of the acid chloride. So that pretty much rules out carboxylic acid derivatives.
Q: On page 780 of the text book, in the mechanism of converting a carboxylic acid to an acid chloride, why does the SO2 group leave instead of Cl? I thought Cl is one of the best leaving groups because it is the conjugate base of a very strong acid.
A: I believe that you are referring to the middle intermediate (in brackets) on the second line of the mechanism. Losing chloride gets you back to the intermediate on the left. This almost certainly happens, but is an unproductive step. That step should probably be denoted as a equilibrium. Losing SO2 would be an irreversible step since it is a gas and this drives the reaction forward to give the acid chloride. Losing a neutral gas is almost always a very favorable driving force for a reaction.

Q: For problem 21.38, is the reason that there is no reactions for addition of cyclohexanol to methyl propanoate because cyclohexanol is a better leaving group than the -OCH3 of methyl propanoate
A: This is not necessarily a straightforward answer. I think the point of the problem is to stress the reactivity order of acyl derivatives; less reactive acyl derivatives can be prepared from more reactive acyl derivatives. For 21.37e, you made cyclohexyl propanoate from the acid chloride and cyclohexanol, which clearly can be done according to the reactivity order; the starting acid chloride is more reactive than the product ester, so the reaction is favored. In 21.38e you are asked if the same reaction can be done starting from methyl propanoate (an ester). Since the starting compound, methyl propanoate, is not a more reactive acyl derivative than the product, cyclohexyl propanoate (they are both esters) you were to conclude that this is not a favorable reaction- so there is no reaction. Your initial thought of looking at the relative leaving group abilities is a very good one. The pKa of methanol is ~16, but cyclohexanol (a secondary alcohol) would probably not be much different (~17), so the leaving group abilities would be roughly the same.
Q: Slide 133 shows the dipole strength of each carbonyl compound. Do these dipole strengths correspond to reactivity?
A: To a degree but no one parameter can explain everything. The prediction that ketones are more reactive than aldehydes which are more reactive than esters holds pretty well. But amides, which have one of the larger dipole are the least reactive.
Q: Are carboxylic acids are the only carbonyl compound reduced by Borane? I know that it is more selective.
A: No, aldehydes and ketones are also reduced and I suspect that an acid chloride would be reduced as well. Amides and nitriles are reduced to the amines. Ester do not react. S you note. the point is that BH3 can do selective chemistry. It reacts very quickly with carboxylic acids, so a carboxylic acid can be selectively reduced in the presence of virtually any other functional groups with BH3 without the need to use a protecting group.
02-26-2006
Q: Regarding problem 21.35 e, is it possible to get 1-butene from butanoic acid by first using LiAlH4 to form an alcahol and then form the double bond with POCl3?
A: That isn't a great way to do it. LiAlH4 (or BH3) is correct for the reduction of butanoic acid to 1-butanol, but primary alcohols are very difficult to dehydrate to alkenes directly. So the way to go would be to convert it to a halide or tosylate followed by E2 elimination. POCl3 works for secondary and tertiary alcohols.
Chapter 20
02-28-2006
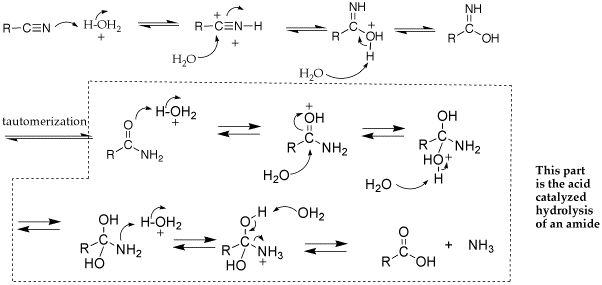
Q: I am trying to draw out the mechanism for the acid-catalyzed reaction of a nitrile to a carboxylic acid, and I'm having a hard time deciding what to do - does the N attack the H30+ and attach the whole thing, does the O from the H30+ attack the C? Then, how will I know if a water comes next to take off a H or if another water/acid attacks the C center. Is there a need to make the C a better nucleophile so it can attack water or the acid?
A: follow the same principles as an acid-catalyed addition to a carbonyl (i.e., acetal or enanime formation and nucleophilic acyl substitution. See mechanism below.
Q: For benzoic acids, I understand that electron withdrawing groups increase acidity by distributing negative charge, but why do they decrease reactivity and vise versa for donating groups?
A: Electron withdrawing groups makes the aromatic ring electron deficient by pulling electrons toward the EWG. This results in the aromatic ring having a partial positive charge character. This can stabilize the benzoate anion (the conjugate base of benzoic acid) through an electrostatic effect. An electron-donating groups makes the aromatic ring electron rich giving it a partial negative charge character. This destabilized the negative charge of the benzoate anion through electrostatic interactions.
02-27-2006
Q: On slide 190, where does the O come from in the final product of the reaction of nitriles with Grignard Reagents?
A: The initial product in the anion of an imine (after Grignard addition). At the end of the reaction, you add water or H3O+ to the reacion and this imine is protonated. In water, the imine will hydrolyze to the ketone plus ammonia. This is the reverse of the imine formation reaction. So the O comes from water
02-26-2006
Q: You mention on slide 185 that LiAlH4 reduces alkyl halides and epoxides. Are alkyl halides (RCH2X) reduced to RCH3 ? For epoxides, is one carbon reduced to a CH while the other becomes a C-OH? Is this reduction selective (i.e., does a certain carbon of the epoxide generally become the alcohol like we saw for acidic/basic hydrolysis of epoxides)?
A: Yes, alkyl halides are reduced to the hydrocarbon and epoxides are opened to alcohols. These reactions are likely to be SN2. For epoxides, the hydride will attack the less hindered carbon to give the more substituted alcohol.
Q: On slide 186, you mention that the carbonyl oxygen of an amide is nucleophilic and can react with electrophiles. Is this not true of all carbonyl compounds (due to the substantial dipole moment of the carbonyl functional group)?
A: Yes, but if you treat a ketone with SOCl2 nothing happens. The dipolar structure explains the reactivity of carbonyl towards most nucleophiles, which add at the C. It also explains that protons (and other strong Lewis acids) will react with a carbonyl at O, however the reactivity of the carbonyl O toward electrophiles is very modest compared to amides.
Q: What would happen if one attempted to dehydrate a secondary/tertiary amide with POCl3 or SOCl2 ? (no reaction?) Is there any way to convert these compounds to carboxylic acids?
Because there is a group or groups on the nitrogen, the dehydration can not proceed unless you break the bond between the N and the group. You end up getting a species know as a Vilsmeier salt (see attached) which is a very powerful electrophile and can be used in F-C acylation reactions. If there is nothing for this salt to react with, it will be hydrolyzed back to the original amide when you add water at the end of the reaction; the ocerall result is no reaction
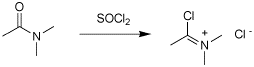
Q: Why is the hydride ion the only nucleophile capable of adding twice to a nitrile? (do all other nucleophiles give the imine? is the imine stable unless hydrolyzed by aqueous acid/base to give the ketone?) I also have in my notes that only hydride ions from LiAlH4 are capable of adding twice to a nitrile. What about BH3?
A: Borane reduces nitriles to amines. I don't know about all other nucleophiles, but since the intermediate (imine anion) from the first addition already has a negative charge, it is much harder to add a second nucleophile to give the di-anion. LiAlH4 is the only reagent reactive enough to add again. Borane is a slightly different case. Because the boron is such a powerful Lewis acid, it is strongly associated with the nitrogen- this bond has a lot of covalent character it it so the initial negative charge is largely neutralized and this allows the second hydride addition to occur.
Q: When a nitrile is reduced with DIBAL to give an imine, will the imine hydrolyze spontaneously to the aldehyde (if in aqueous solution), much like the product of Grignard addition to a nitrile spontaneously hydrolyzes to give a ketone? (does this mean that carbonyls are more stable than imines?)
A: Yes. When aqueous acid is added at the end of the DIBAH reduction or Grignard reaction, a imine that is formally derived from an aldehyde and ammonia, is obtained. This spontaneously hydrolyzes in aqueous solution to give the aldehyde and ammonia. I am not sure about stability (i.e., bond energies, etc.) but there are other considerations. The nitrogen of an imine is basic and is easily be protonated, even at neutral pH. The gives a positively charges iminium ion to which water will added very rapidily. In aqueous solution, the equilibrium is pushed to aldehyde and ammonia.
Q: If a set of atoms are conjugated, must each one of these atoms have the same hybridization?
A: Not necessarily; a C-C triple bond can be conjugated with a carbonyl, a nitrile can be in conjugation with an aromatic ring. The hydridization of the atoms must be such, that allows for an overlapping array of p-orbitals. So for an amide, the hybridization of the nitrogen is sp2 because this allows the lone pair of the nitrogen to be in a p-orbital that can conjugate with the pi-bond on the carbonyl (which are also in p-orbitals). This also true of the nitrogen of pyrrole and of an enamine (see below).
Q: You mentioned that NaBH4 is more basic than LiAlH4 and will undergo acid-base chemistry with carboxylic acids. What accounts for the basicity of NaBH4 (as opposed to lithium alumnium hydride)?
A: Any anionic species has the potential to be a base. Both are NaBH4 and LiAlH4 are basic reagents and the first mol of hydride from either will deprotonate the acid to give a carboxylate. NaBH4 is not a strong enough hydride source to react with the carboxylate further. As noted above with regard to the nitrile reduction, LiAlH4 (which is in an aprotic solvent) is reactive enough to reduce the carboxylate ion of the acid.
Q: Why are aldehydes and ketones more downfield in 13C NMR than carboxylic acids and their derivatives?
A: As we have noted the C=O bond has a large dipole moment and the carbon is deshielded as a result. For aldehydes and ketones, the alkyl groups or -H do not compensate much in terms of electron donation (which will shield the nuclei) for the large pull of the carbonyl oxygen. Carboxylic acid derivatives where the Y group can be OH, OR, NR2 or -Cl have lone pairs of electrons and can donate electron density though resonance; thus, the carbonyl carbon is not as electron deficient and consequently not as deshielded. Also, each of these group are electronegative in their own right. The individual bond dipole of the -Y group to a degree opposes the C=O bond dipole. Thus, the carbonyl carbon bond dipole is reduced and the carbon is less deshielded.
Nitrile are much more shielded for a different reason. If you remember the cryptic explanation of ring currents for aromatic rings that cause aromatic protons to be unusually deshielded, but a proton in the middle of the ring would be unusually shielded. There is a similar explaination for triples bonds. There is a ring rurrent for triple bonds that shield the carbons of nitriles and acetylenes which is why 13C and 1H resonances of these compounds are upfield compaied to alkenes.
Chapter 19
02-28-2006
Q: Ketone Hs on an alpha C don't couple to H across C=O group, right?
A: That is correct, the alpha protons on one side of the carbonyl do NOT couple with the alpha protons on the other side of the carbonyl.
Q. Is slide 164 showing that the formation of the beta amino aldehyde/ketone is thermodynamically favored over the formation of the imine?
A: yes, conjugate addition of an amine is favored over imine formation for alpha,beta unsaturated ketones and aldehydes.
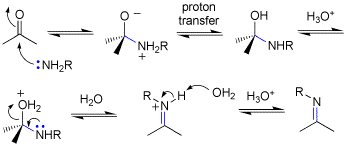
Q: The imine formation mechanism that was drawn on the board had a H on the O in the second step without any explanation of how it got there. The book mentions a proton transfer but our drawings kept the NH2R intact until a water removed one H in the third step. Did we have one too many steps with that H in our reaction?
A: See attached. The book, and I abbreviated the mechanism slightly. The prton is tranbsfered from N to O-, and this can be drawn as solvent or some conjugate base mediating the transfer as we have been drawing for other mechanisms.
Q: Slide 151 mentions that the mechanism for enamine formation includes tautomerization but I don't see where that step comes in in the reaction shown on p. 699?
A: I mis-spoke, it is not a tautomerization. It is loss of a proton followed by rearrangment of the imine to an enamine. It is the last step, struture second to the bottom going to the bottom structure.
Q: Is the reduction of an ester to an aldehyde not a reliable reaction?
A: It is not a reliable reaction in the lab, but you can use on paper.
Q: In the reverse of the base-induced nucleophilic addition reaction to form a 1,1-diol, the second step shows the reformation of the C=O bond through the expulsion of the OH. I didn't think that -OH was a good enough leaving group to be able to be expelled in this manner?
A: Normally it is not, but under basic conditions it there are no protons available to protonate it. Since this is cone under basic cinditions, this step generates the base catalyst and the driving force is the bond energy that is gained from re-forming the C=O.
Q: Is it CrO3 or Cr(VI) that will prepare ketones from secondary alcohols?
A: Ether PCC or CrO3/HCl will give ketones from secondary alcohols. The difference
is for primary alcohols. CrO3/HCl converts them to carboxylic acids while PCC
oxidizes them to aldehydes
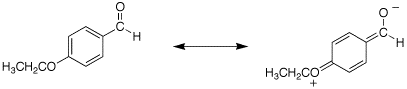
Q: The sprctra for problem 19.67c are for p-ethoxybenzaldehyde. The IR for the carbonyl is listed as 1695 cm-1. Is this correct? It does not really match that of a conjugated aldehyde which should be 1705 cm-1.
A: The carbonyl of p-ethoxybenzaldehyde really is at ~1695 cm-1. I think this is because the aromatic ring is so electron-rich due to the p-ethoxy group and an very strong contribution of the resonance form shown below.
Q: The Ylide in the Wittig reaction will only reacts with aldehyde or ketones but as far as the limitations on the reaction, are alcohols amines and carboxylic acids not tolerated in the ylide formation or the ketone or aldehyde or all? (Can you have them present anywhere?)
A: It would be a good idea not to have potentially acidic functional groups in either the ylide or reactant. Those include carboxylic acids, alcohols and amines. Esters and amides are OK but acid chlorides and anhydrides would not be.
Q: For the conjugate addition of alkyl groups via Gilman reagents, I was wondering if the Gilman's reagent is limited to alkyl groups. I know that we said alkynyl groups don't work in Gilman's Reagents, but what about something like (HOCH2CH2)2CuLi ? Can functional groups such as an alcohol be added through this reaction or is it strictly alkyl groups?
A: Gilman reagents can be alkyl, vinyl or aryl.
Any basic reagent such as Grignards, Gillmans and Wittigs are going to have compatibility issues with acidic functional groups either as part of the reagent or the substrate it is reacting with. Acid base chemistry will be much faster. The key to all of these reagents is that they are negatively charged (and hence, their basic nature) nucleophiles. If they do acid base chemistry, their charge and nucleophilicity are gone. So the Gillman reagent you wrote above wouldn't fly because of the hydroxyl group- you would not be able to generate the organilithium species. If a Wittig reagent has a ketone or aldehyde as part of the ylide, it could react with itself rather than with the substrate.
Q: I had written in class that the Wittig reaction is not compatible with acidic groups such as alcohols. Just to be sure, this means that if an alcohol were on the same molecule as the ketone reactant, the Wittig reaction wouldn't take place?
A: Yes, the negative charge of the ylide would be re-protonated by the acidic functionality. The ylide and ketone/aldehyde substrate should be free of acidic protons. The ylide should probably not have aldehyde or ketones either since the Witting reagent would react with itself, unless this is by design.
02-27-2006
Q: The equilibrium for hydration of aldehydes and ketones favors the carbonyl compound (except with the least hindered carbonyls such as formaldehyde and acetaldehyde). Because of this, is acid or base catalyzed hydration an acceptable reaction for the synthesis of gem-diols? If not, what other methods do we know to synthesize a gem diol?
A: This is something of a model or a prototype reaction for the nucleophilic addition to carbonyls. Hydrates of ketones and aldehyde are intermediates for some reactions, i.e., Jones oxidation. Gem-diols really can not be synthesized. You put and aldehyde or ketone in water and you get an equilibrium with the hydrate. You remove the water and you get the carbonyl back.
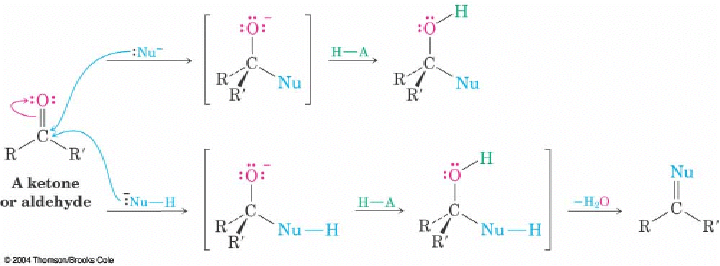
Q: On page 689 the book shows 2 different variations that a nucleophilic addition reaction can follow. What determines which variation the reaction will take? Is it the presence of a hydrogen on the nucleophile?
A: The nature of the nucleophile. The top reactivity is for hydrides and most carbon anion like Grignard reagents. The chemistry is pretty much limited to the addition step and an alcohols is usually the product. The bottom reactivity is for nucleophiles that have additions lone pairs of the electron or a proton that can be lost after the initial addition (the loss of the protons unveils a lone pair of electrons). In these cases, the lone pair can puch out the hydroxyl group (usually after protonation as H2O) to give a C=Nu. These nucelophiles have either O or N as the nucleophilic atom. For N, the C=N- is an imine and is stable or can rearrange to an enamine. Of O, the C=O-(+) is not a stable end product but is an intermediate for another nucleophilic addition (i.e. acetal formation)
02-26-2006
Q: Do you know why aldehyde/ketone hydration is more favored for less substituted molecules (e.g., formaldehyde)?
A: This probably has to so mostly with sterics.
Q: Can one control the direction of the hydration equilibrium?
A: If you remove the water, the carbonyl is produced. I don't think that you can push the equilibrium toward hydrate.
Q: Are there any other nucleophilic addition reactions of H-Y that are favorable besides cyanohydrin formation?Do you know why does cyanohydrin formation favor the cyanohydrin?
A: Not that I can think of. Cyanohydrin formation is pretty unique.
It defies conventional wisdom because H-CN is a stronger acid that either water or an alcohol, so you would predict that the equilibrium would not favor cyanohydrin formation. However, I think the driving force must be the added energy of the new C-C bond that is being formed. Interpreting equilibrium can be tenuous because very small energy differences can significantly tip the balance of an equilibrium. An energy difference of zero means a 1:1 mixture of products and reactants. A change of just ~14 KJ/mole (which is a very small amount of energy) change the equilibrium to > 99.9 : 0.1.
Q: Are carbinolamines stable in the absence of catalyst?
A: I think you mis-spoke here. As you know, the catalyst only affects the rates of formation, not the stability of the products.
Some carbinolamines are stable and observable. Usually, there is some structural effect that holds it together, like being constrained in a ring.
Q: What is implied by the isoelectronicity of an imine with a carbonyl? That the two should exhibit very similar chemistry? Enamines are isoelectronic with enolate anions? Does this imply that the two also have similar chemistry?
A: Yes. The imine is in the same oxidation state as a carbonyl and the bonding arrangement of a C=O and C=NR are similar. So there is overlapping chemistry. A Grignard reagent will add to an imine to get an amine; they can be reduced to amines, the alpha-protons are acidic, etc. However, there is hydrolytic chemistry of imines which often competes with these reactions, which is not similar to a ketone or aldehyde
We will in the next Chapter 23 that enamines are used a enolate equivalents.
Q: Is the "alpha nitrogen" of a hydrazone (such as those generated in the Wolff-Kishner reaction) acidic (due to the neighboring imine group)? Would the acid catalyst from the previous step (hydrazone formation) have to be neutralized first? Would one just add excess base?
A: The hydrazone N-H is much more acidic than a normal N-H. There is a pH range where hydrazones can be formed. I believe that the optimal pH is just on the acidic side of neutral. But just like imines, hydrazone can be formed at basic pH, although the rate is slower (the book mentions this, but I thought that the discussion was somewhat opaque and tmight cause more questions than clarity). So the W-K reduction is most often done without a discrete acid-catalyzed formation of the hydrazone step. McMurry presents the reaction as simply hydrazine and KOH and my notes did as well. This is somewhat inconsistent with the previous acid catalyzed formation of imines, however.
Q: Are there no other functional groups that are reduced (or interfere with) the Wolff-Kishner reaction?
A: Groups that can not tolerate strongly basic conditions would not be compatible with the W-K reduction. For instance, a ester would be hydrolyzed, alkyl halides would eliminate, etc. However, these are reduction conditions that unique for ketones and aldehydes. Almost any other reducable group will not be affected by the W-K reduction, unless they were hydroxide sensitive (so an acid chloride is going to be hydrolyzed).
Q: On slide 157, a ketone is selectively reduces in the presence of an ester with NaBH4. Am I correct in saying that this would still work if the ester were a carboxylic acid instead? While the ketone would again be converted to a ketal, the acidic conditions would cause the carboxylic acid to be converted to an ester by a Fischer esterification reaction. This ester could then be reduced with LiAlH4 and the protecting group could be removed with aqueous acid. Would this be a viable way to selectively reduce a acid?
A: NaBH4 does not reduce esters, carboxylic acids, amides or nitriles. It depends on the reaction conditions, however, if the substrate on that slide were a keto-acid, I believe that you would get the acid out at the end of the reaction rather that the ester of ethylene glycol. This reaction is done in a Dean-Stark trap with a stoicheometric amount of ethylene glycol and benzene as the solvent. The Fischer esterification generally requires huge excesses of the alcohol to drive it forward. So the ketalization reaction would be faster because the ketone is more reactive. Ethylene glycol esters would be minor products of the reaction.
We noted in the Chapter 20 that borane is a method for selective reduction of a carboxylic acid. So al alternative to that shown in Slide 157 is to hydrolyzed the keto exter to a keto acid and treat with borane. This would give the keto alcohols.
Q: Why can't tetrasubstituted alkenes be formed in the Wittig reaction (the book only goes so far as to say "steric reasons" are involved)? Is it unwise to add a disubstituted triphenylphosphine to an aldehyde to give a trisubstituted alkene? The book mentions the reaction does not work well with disubstituted CR2 groups.
A: Sterics are the issue. The mechanism of the Wittig reaction is actually a little complicated. It turns out that the initial addition of a Wittig reagent to the ketone or aldehyde is actually reversible. This is in contrast to the addition of other carbon anions such as Grignard's, etc., where the addition is irreverible. So the collapse of the oxaphosphatane to the alkene and Ph3PO is the "committed" step. If the formation and collapse of the oxaphosphatane is slow relative to the reverse of ylide addition, there will be no reaction. For a tetrasubstitute alkene, there can be considerable steric strain because both sets of cis-groups on the alkene would be "eclipsed," so depending on the steric requirements of these groups, collapse of am oxaphosphatane to a tetrasubstituted alkene is likely to be unfavorable. McMurry does not mention any of this an I believe that it is an unnecessary details.
Q: Are there any limitations on which molecules can be used as Wittig reagents (besides any compound containing an acidic group)?
A: It would not be a good idea to have a ketone or aldehyde as part of theWittig reagent as they can then self condense.
Q: Why is the carbonyl carbon resonance in 13C NMR so weak? Doesn't the intensity depend on the magnitude of the dipole moment as in 1H NMR? If so, wouldn't one expect this resonance to be strong?
A: This is really beyond the scope right now and the deeper reasons are well beyond my understanding. Almost all textbooks described NMR the way it was done 30 years ago. The NMR instruments do not work that way anymore and the physics and math required to fully understand NMR are bewildering (very advanced, at least for me).
The book very briefly mentions FT-NMR (Chapter 13.4) which is the way NMR instruments work. The dipole of the nuclei does't play a significant role in NMR. NMR is related to the interaction of the magnetic moment of a nuclei with an external magnetic field. Not all nuclei are NMR active. In any event, the magnetization of the nuclei is perturbed by a pulse of EM energy. The nuclei react to this perturbation and this gives rise to the NMR signal. In FT- (or pulsed or time-domain) NMR this perturbation is done many times and a spectrum is collected each time. The signals are then averaged (this is called signal averaging) and converted into the spectrum. After the nuclei is perturbed, it relaxes back to it original state; this information is collected and is one spectrum; it is then perturbed again after a time delay to allow for the relaxation and this is how multiple spectra are collected and averaged. All protons relax quickly and over a fairly uniform time range, in a few milliseconds or less, so the spectra can be collected very quickly. Carbons however relax slowly relative to protons and over a large range of the relaxation rates. Most carbons relax within a second or so, but a carbonyl can take several seconds or longer to relax and this is difficult to predict. So while the data is being collected, the different rates of relaxation cause the intensities of the carbon signals to differ. The delay between pulses is often not set sufficiently long enough to allow full relaxation and this added to weak signal of the carbonyl. So carbon intensities do not reflect the relative number of nuclei giving rise to that signal and therefore can not be reliable integrated without special techniques. Because carbon is such an insensitive NMR nuclei, the intensity is sacrificed in order to obtain as many pulses as possible. While the data for a proton spectrum can usually be collected in less than a minute, carbon spectra can take hours to collect.
Q: How does one determine the oxidation state of an organic compound? For example, you mentioned that ketones and ketals are in the same oxidation state, but how would one determine this just by looking at the structures? I would imagine it involves counting the number of bonds to atoms of equal electronegativity, lesser electronegativity, and greater electronegativity and taking the sum of this along with the formal charge on the atom would give a rough indication of its oxidation state. Is this at all correct?
A: For organic compound it is really much simpler than that. A carbon has a valancy of four and we are concerned about the oxidation state of a specific carbon or functional group. A hydrocarbon is in the lowest oxidation state. The oxidation state increase when a C-H bond is replaced with a more electronegative atom or group (which is almost anything). Decreasing the hydrogen content of the molecule also counts as increasing the oxidation state. Thus, a pi-bond counts as an increase in oxidation state so an alkene would be in a higher oxidation state than a alkane, but is that same as an alcohol or alkyl halide. This has to be correct if you consider the hydration of an alkene or addition of HX to an alkene. These are not redox reactions. If you were to say the alkene is being oxidized upon hydration, there must be another product which is being reduced, which there is not. The conversion of a carbonyl to an acetal/ketal is similar to the hydration of an alkene. So the acetal/ketal carbon (which was formally the carbonyl carbon) has not changed oxidation. Slide 88 shows the oxidation the oxidations of the compounds we have discussed.
Q: Could you please explain why aromatic aldehydes are less reactive than aldehydes? Does the same reduction in reactivity apply to aromatic ketones as well?
A: Both aryl aldehydes and ketones are less reactive than aliphatic aldehydes and ketones because the aromatic ring donates electron density to the carbonyl through resonance (see page 691), This donation makes the aromatic ring electron deficient and the partial positive charge is delocalized into the ring. The reduced reactivity pertained to nucleophilic addition, since the carbonyl carbon is less electropositive in this case.
02-25-2006
Q: Do imines and enamines have synthetic usefulness? I recall you saying that imines are of crucial importance in biochemistry. Can imines or enamines be converted to nitriles or amides? Can imines be reduced to amines with lithium alumnium hydride?
A: Imines are generally used as precursors for amines. We will cover a reaction called a reductive amination in which a ketone or aldehyde is reacted with an amine under reducing conditions (such as H2, Pd/C). The amine and ketone form an iminium ion (protonated imine), which is very easily reduced to give an amine. This is one of the best ways for making substituted amines. We will see in the next chapter that enamines are the equavalent of enolates. Oximes, which are related to imines can be dehydrated to nitriles.
In biochemistry, an imine is referred to as a Schiff base. The reactions of an entire class of enzymes that are dependent on the coenzyme pyridoxal phosphate, go through a critical imine linkage between the coenzyme and the substrate. As also in the class of retinal binding to opsin, imines are often transient linkages between a receptor and a protein or enzyme and substrate.
Q: How does the chemistry of organolithiums compare to that of Grignard and Gilman reagents? The text frequently discusses the latter two but rarely mentions organolithiums.
A: I probably should just stop mentioning organolithium reagents (I guess I don't always remember what is in the first half of the text). In general, the reactivity of a Grignard's parallels that of an organolithium (organolithiums are actually more reactive). Gillman reagents are somewhat less reactive, but they possess unique reactivities that other organometallic reagents don't have. The coupling reaction with an acid halide being one of them.
Q: DIBAL allows partial reduction of esters to aldehydes. Does it partially reduce any other functional groups?
A: Lactone which are cyclic esters are reduced to aldehydes. In this case the product has an appended alcohol group, which comes from the other oxygen of the lactone. Nitriles are reduced to imine by DIBAH, which hydrolyze to aldehydes. DIBAL also reduced aldehydes and ketones and is used for many types of reductions.
Why are acidic groups not tolerated in the Wittig reaction?
Since the reactive form of the Wittig reagent is the ylide, the negative charge
can potentially do acid-base chemistry with acidic groups. If the ylide is re-
protonated, then the phosphonium salt results.
Q: Aryl ketones are reduced to methylene groups via hydrogen over palladium. Does this reaction also work with benzaldehyde?
A: Yes
Q: What is the mechanism for heat-induced expulsion of water from carboxylic acids to form an anhydride?
A: The mechanism I would write is one eqivalent of acid protonates a second equivalent to give a RC(=OH+)-OH and a carboxylate ion. Now draw the acyl substitution reaction as we did today for other reactions, to give the anhydride and water as the product. The reaction must be done at a temperature were the water is driven out of the reaction. If water is present, the water will simply add to the anhydride and the reverse reaction will proceed.
Q: Do anhydrides react with Grignard reagents? (it is not listed as such on page 788 of the text in Figure 21.7)
A: Yes they do. Grignard addition will give the tertiray alcohol just as for acid chlorides and esters. For the most part, anhydrides are used interchangably with acid chlorides (except for the reaction with Gillman reagents). The book notes, however, that an anhydride is actually an inefficient reagent in that two mols of the starting acid are used to make the anhydride, but if you react an anhydride with, say an amine to give an amine, only half of the anhydride reacts. The other half is lost as the carboxylate. That is OK for simple anhydrides such as acetyl anydride. However, if you wanted to make an amide from a carboxylic acid, but the carboxylic acid had to be synthesized from other starting materials, you would not want to make an anhydride out of it before reacting it with an amine. The acid chloride would be the way to go.
Q: Why is the NH2 group more nucleophilic than the OH group? Also, in the synthesis of aspirin, why does the alcohol -OH of salicylic acid rather than the carboxyl -OH react with acetic anhydride?
A: I am not sure I have a good answer for that. In general, amino groups are much more reactive than alcohols toward electrophiles. When an neutral amino group reacts with a electrophile the initial product has a positive charge on nitrogen. Nitrogen handles having a positive change very well as there are many stable ammonium salts. While oxygen can handle positive change transiently, there are really not many stable species that have a positive change on oxygen. Also, if you consider what makes for a good nucleophile (Chapter 11), good nuclephiles are the conjugate bases of weak acids (i.e., strong bases). The pka of the ammonium ion (NH4+) is about 10, while the pKa of H3O+ (a surrogate for ROH2+) is -1.7.
Q: Is the basicity of an amine comparable to the acidity of an alcohol?
A: The pKa of ammonium ions are in the 9-10 range. Most alcohols are in the 16-18 range. So there is not much acid base chemistry between an amine and a normal alcohol.
Q: Why does the book say that the conversion of acid chlorides to esters is "more general" than the conversion of acids to esters? The text claims that the Fischer esterification reaction is limited to the synthesis of methyl, ethyl, propyl, and butyl esters because of the need to use excess liquid alcohol as solvent. Why is this limiting?
The Fischer esterification is practically limited to making esters with simple alcohols because they are used a solvent. Also, as the alcohols get larger, the boiling point gets higher, so there is a practical consideration of how to get the product out of the reaction. If you wanted to make an acetate ester of an elaborate alcohol that required many steps to synthesize, you couldn't practically use it as solvent for the Fischer esterification with acetic acid (since acetic acid would be the limiting reagent in that case). So the more efficient way to go would be to use your fancy alcohol as the limiting reagent and react it with an excess of acetyl chloide (or anhydride) in pyridine solvent.
Q: The acid chloride reduction by lithium alumnium hydride proceeds through an aldehyde intermediate before the primary alcohol is produced. Can the aldehyde be isolated with DIBAL at -78 degrees celsius? (out of curiosity, why is -78 degrees celsius often used as a reaction temperature in the book? Is this because the freezing point of CO2 (dry ice) is around this temperature and therefore is a particularly easy temperature to attain?)
A: No. With DIBAH and an ester, the intial addition of a hydride give an tetrahedral intermediate with a negative charge on oxygen. The Al of DIBAH is coordinated to this oxygen. This Al is also coodinated to the othe oxygen of the ester linkage. The bi-dentate coordination is critical at stabilizng this intermediate. if it breaks down, OR is expelled and the aldehyde is generated which will react fruther with more DIBAL to give the primary alcohol. The Al-coordinated intermediate is still in an aldehyde oxidation state and in fact can be viewed as a acetal of sorts- it does not react with nucleophiles. For an acid chloride, this intermediate does not have sufficient stability partly because chloride is such a good leaving group and partly because -Cl is not as good a ligand for Al as oxygen. So the aldehyde is generated as an intermediate in the reaction and is further reduced. When LiAlH4 is used, the nature of the Al is much different than in DIBAH, so the coordination phenomena I described above does not occur.
When you powder dry ice and add isopropyl alcohol, the temeperature of the resulting slush bath is -78° C.
Q: Can the Grignard addition to esters be paused at the ketone stage?
A: In general no. The ketone intermediate is actually more reactive toward nuleophilic addition than the starting ester. The the second equv. adds faster than the first.
Q: For the reduction of amides to amines, why does the oxygen (O-) leave instead of the amine group (as occurs in reductions of other carboxylic acid derivatives)? Is the O- a better leaving group than an amine?
A: Yes, but with LiAlH4 it is not a simple alkoxy group, it is the oxygen coordinated to the aluminium of LiAlH4. It leaves to form some aluminium oxide or something. For the reduction with borane, the oxygen leaves are a borate.
02-23-2006
Q: Could you go over problem #27 from chapter 19?
A: The geometry of the nitrogen atom is trigonal planar and it is sp2 hybridized.
The lone pair of electrons on the nitrogen are in an unhybridized p-orbital.
In the geometry shown, all carbons and the nitrogen are (or nearly are) co-planar. This geometry allows for the lone-pair of electrons on Nitrogen to beconjugated with the pi-electrons of the C=C. This is a stabilizing interaction.An important resonance structure of enamines is where there is a negative chargeon the carbon and a N=C double bond. We will see this is Chapter 22.
02-22-2006
Q: At the bottom of page 689 in the book, for the second variation > of nucleophilic addition to a ketone, why does the -OH leave. I thought -OH was a bad leaving group. Does the proton transfer occur from the nucleophile to protonate the -OH?
A: This is acyually an abbreviated mechanism. Note that the -OH actually ends up as H2O. There is a proton transfer for the -Nu-H to the -OH to give -OH2 as the leaving group. This is better shown in Fig. 19.8 (page 687) for the mechanism of imine formation
Q: In the mechanism for the Grignard reaction (p695), why does water remove the MgX?
A: The -O(-) MgX+ is more or less an ionic bond. When water or H3O+ is added at the end of the Grignard reaction the -O(-) is protonated to give the -OH, which is a covalent bond. The simple answer is that the overall bonding is energetically more favorable.
Q: I don't understand why conjugate 1,4 addition occurs? It seems that whenever a carbonyl is present, the carbonyl carbon will always be more polarized and bear more of a partial positive charge than the Beta carbon, so why does the nucleophile attack the beta carbon to begin with?
A: This is a very complicated issue. The nature and reactivity of the nucleophiles figure into the equation as well and I think that this is beyond the scope of the course at the moment. It is based largely on experimental observations that have been formulated into a set of empirical rules rather than a theoretical analysis. It is known as hard-soft acid-base principles which takes into account the solvent conditions, the counterion and the nature of the nucleophiles and electrophiles.
To summarize the observation for conjugate (1,4-) addition versus direct (1,2-) addition:
All organometallic reagents, organolithiums, Grgnard reagents and diorganocopper reagents, will undergo direct addition to aldehydes including alpha,beta-unsaturated aldehydes.
Organolithium and Grignard reagents undergo direct addition to ketones, including alpha,beta-unsaturated ketones.
Diorganocoper reagents undergo conjugate addition to alpha,beta-unsaturated ketones, but direct addition to alpha,beta-unsaturated aldehydes.
The same thing, perhaps a little simpler: Organolithium, Grgnard reagents and diorganocopper reagents undergo direct addition of aldehydes and ketones, except for the reaction of diorganocopper reagents with alpha,beta-unsaturated ketones which results in conjugtaed addition.
Amines undergoe conjugate addition to alpha,beta- unstaurated ketones and aldehydes.
Q: Finally, in 19.16 the book goes over where different aldehydes and ketones absorb on the IR spectrum. How important is it that we know these differences and what are some general guidelines determining the absorption patterns of different ketones and aldehydes?
A: It is hard for me to answer that. The book often presents this as if you need to determine a structure only by IR. In general, no one piece of spectral data can determine the structure of a molecule conclusively. Structures are determined using a combination of spectra information. Aldehydes have a distinct chemical shift in the 1H NMR spectrum. The specific location of the C=O strtch in the IR can be very helpful in determining if the aldehyde is conjugated (to either a C=C or aromatic ring). So knowing that the typical IR stretch for an aldehyde C=O is 1730 cm-1 and that conjugation moves this number down to ~1700 is useful. The same for a ketone C=O.
I would say, that knowing the standard stretches for an aldehyde (1730), ketone (1715) and ester (1735) would be beneficial and that conjugation moves these the lower wavenumbers. For cyclic ketones, small ring sizes move the C=O stretch to higher wavenumbers. Aldehydes/ketones can be differentiated from acid/esters/amides by the 13C chemical shifts of the carbonyls.
02-17-2006
Q: On slide 159, is the Wittig reagent supposed to be the phosphonium salt or the ylide because CH2 is added, not CH3?
A: That scheme shows the reagents needed to generate the ylide. So showing the phsophonium salt plus a strong base, in this case methyllithium, indicates that the ylide is being formed. Methyl lithium deprotonates the phosphonium salt to give the ylide.
Q: On slide 160, why does the alkene form in that position? Also, why does the reagent change again? - now phosphorane
A: I will change the phosphorane to the ylide in the notes and re-post them. I think that the reactivity is better understood using the ylide form. This is a very good point.
The position of the alkene from a Wittig reaction is completely defined by the reactants and is an important feature of the Wittig. It forms between thecarbonyl carbon and the anion of the ylide. If you look at the mechanism on slide 159, the alkene forms between the green carbon (the anion of the ylide) and the blue carbon (the carbonyl carbon)
Q: Between the ylide and phosphorane shown on slide 158, does one resonance form predominate?
A: I don't know if one resonance form contributes more than the other to the resonance hybrid. Keep in mind that that neither is the actual structure. I would say that it is a personal preference to denote the ylide or phosphorane. They are equivalent, although the ylide probably more acturatly depicts the reactive species.
Q: Regarding the cyclic acetal shown on slide 157, what is the hybridization of the C attached to both oxygens?
A: The carbon of any acetal or ketal is Sp3 hydridized. However, it is still in the same oxidation state as a ketone or aldehyde. On slide 88 (Ch. 17) there is a diagram that shows increasing oxidation state. The geminal dichloride is the same as a ketone/aldehyde and imine. The oxidation state of the acetal/ketal or hydrate is the same as the geminal dichloride. However, acetals and ketals do NOT have the same reactivity as the carbonyl
Q: On slide 155, can any strong base (NaOH or KOH) be used in the Wolff- Kishner reaction?
A: KOH is interchangeable with NaOH for the W-K reduction
Q: On slide 154, is it a base that adds to the iminium intermediate to convert it to the enamine form?
A: Yes. Water can act as a base. In this examples the acid that was used was H3O+, so H2O is the conjugate base.
Q: What does the phrase on slide 152 "imines are isoelectronic with a carbonyl" mean?
A: The C=N double bond of an imine has similar bonding properties with a C=O of a carbonyl and consequently they have similar reactivity. We will see some reactions of C=N later. Imines can be reduced (with hydride) and reacts with nucleophiles such as Grignard reagents to give amines in an analogous fashion as addition reactions to carbonyls.
Q: On slide 147, the reaction shows a ketone, but you can also hydrate an aldehyde? Are base or acid catalyzed reactions occur at the same rate when you have the choice as shown on slid 147 (with one having the hydroxide as a better nucleophile and the other having the protonated carbonyl as a better electrophile)?
A: The carbonyl in the substrate were not meant to be specific, so it can be either a ketone or aldehyde. This is the way the text denoted a general carbonyl compound. In fact, aldehydes hydrate much more readily than ketones.
Q: On slide 143, as the last reaction is written using the Gilman's reagent, is this an SN2 reaction? If the object to make a ketone, why did our notes show production of a benzene with R-group or an alkene with R group attached?
A: No, this is not an SN2- SN2 reaction only occur when the whe the C-X has an Sp3 carbon. I don't thing the mechanism is straigntforward for this reaction but is may be a nucleophilic acy substitution, which will be covered in Chapter 21.
The "R" denotes any organic groups and is meant to show that the reaction is general. The top of slide 143 is the Friedel-Crafts acylation which is limited to the preparation of aryl ketones. The reaction of an acid chloride with a Gilman reagent is a much more general way to make ketones, includinh a,b-unsaturated ketones and aryl ketones. Just about any acid chloride can be used, and we subsequently said that the Gilman reagent can be any have alkyl groups, vinyl groups and aryl groups (but not alkynyl). So this reaction can potentially make a ketone with a wide variety of groups on either side of the carbonyl depending on the starting acid chloride and Gilman reagent (which can be made from alkyl, vinyl and aryl halides)