McLean Research Group Announcements
 |
Vanessa Allwardt et al. Receive a Highly Cited Paper Award for their Organ-on-a-Chip Review in Bioengineering:10.12.2022 “Translational Roadmap for the Organs-on-a-Chip Industry toward Broad Adoption” Vanessa Allwardt, Alexander J. Ainscough, Priyalakshmi Viswanathan, Stacy D. Sherrod, John A. McLean, Malcolm Haddrick, and Virginia Pensabene, Bioengineering 7(3), 112, 2020. |
 |
Group Members Present at the 2022 ISIMS Conference in Memphis, TN07.24.2022 The McLean Research Group members presented their research at the 2022 International Conference on Ion Mobility Spectrometry in Memphis, TN: Poster Presentations: Madelyn James “Ion Mobility-Mass Spectrometry Structural Mapping of Discrete Mass Polyurethane Oligomers” – Outstanding Poster Award! |
 |
The McLean Research Group Members Present their Work at the 2022 ASMS Conference in Minneapolis, MN06.04.2022 The McLean Research Group members presented their research at the 70th annual American Society for Mass Spectrometry (ASMS) Conference, which was held in Minneapolis, MN: Oral Presentation: David Koomen “Untargeted Metabolomics Strategy to Identify Unknown Phase II Anabolic Androgenic Steroids with Liquid Chromatography-Ion Mobility-Mass Spectrometry for Doping Control Workflows” Poster Presentations: Jody C. May, “Implementation of Temperature Induced Protein Unfolding on a Drift Tube Ion Mobility-Mass Spectrometer” |
 |
Congratulations to Dr. Bailey S. Rose on a Successful Dissertation Defense!03.23.2022 Dr. Rose’s dissertation is titled “Multidimensional Analyses for High Confidence Untargeted Molecular Annotation using Structurally Specific Ion Mobility-Mass Spectrometry”. We’re proud of you Bailey! |
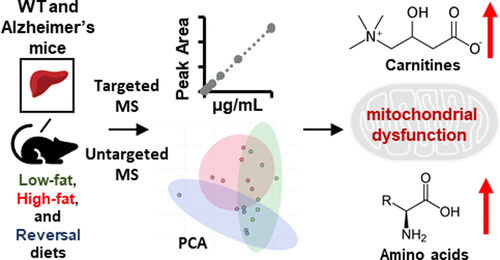 |
Congratulations to Amelia Taylor and co-authors on their recent publication!01.25.2022 “Targeted and Untargeted Mass Spectrometry Reveals the Impact of High-Fat Diet on Peripheral Amino Acid Regulation in a Mouse Model of Alzheimer’s Disease” AL Taylor, DE Davis, SG Codreanu, FE Harrison, SD Sherrod, JA McLean; Journal of Proteome Research 20(9), 4405-4414 (2021). Amelia Taylor is an undergraduate researcher in the group and her first-author publication focuses on finding dysregulated metabolites in an Alzheimer’s mouse model. |
 |
Prof. McLean elected as a Fellow of the National Academy of Inventors for 202112.08.2021 From the NAI press announcement: “The NAI Fellows Program highlights academic inventors who have demonstrated a spirit of innovation in creating or facilitating outstanding inventions that have made a tangible impact on the quality of life, economic development, and the welfare of society. Election to NAI Fellow is the highest professional distinction accorded solely to academic inventors.” |
 |
The McLean Research Group Members Present their Research at the 2021 ASMS Conference in Philadelphia, PA10.31.2021 The McLean Research Group members presented their research at the 2021 American Society for Mass Spectrometry (ASMS) Conference, which was held in-person in Philadelphia, PA: Oral Presentations: John A. McLean, “Refining the Landscape of Lipidomics with High Resolution Ion Mobility (SLIM)-Mass Spectrometry” Poster Presentations: Bailey Rose “Lipid Collision Cross Section Calibration Strategies for High Resolution SLIM-based Traveling Wave Ion Mobility Separations” |
 |
Congratulations to Dr. Berkley Ellis on a Successful Dissertation Defense06.09.2021 Dr. Berkley Ellis’ dissertation (defended June 2021) is titled “Accelerating Natural Product Discovery and Synthetic Biology Workflows Using Multidimensional Molecular Phenotyping”. |
Amelia Taylor and Sael Sohni Receive Undergraduate Research Awards04.28.2021 Undergraduate researcher Amelia Taylor receives the Merck Index Award and the Outstanding Chemistry Research Award, and undergraduate researcher Sael Sohni receives the Department’s Analytical Chemistry Award. Congratulations! |
  |
Congratulations to Dr. Don Davis and Dr. Rachel Harris on Their Successful Dissertation Defense03.23.2021 Dr. Don Davis’ dissertation (defended March 2021) is titled “Multidimensional Separations in Complex Biological Matrices Utilizing Chromatography, Ion Mobility, and Mass Spectrometry.” Dr. Rachel Harris’ dissertation (defended March 2021) is titled “Analysis of Biopolymers Using Structurally Selective Analytical Techniques in Combination with Mass Spectrometry”. |
Welcome Hawkins, David and Allison to the Research Group!01.01.2021 |
John McLean Leads Arts and Science Grand Challenge Initiative08.26.2020 Prof. McLean leads the recently launched Advancing Solutions to Climate and Ecological Needs and Discoveries (ASCEND) Initiative. It is one of six Grand Challenge Initiatives supported by the College of Arts and Science. |
  |
Congratulations to Dr. James Poland and Dr. Jaqueline Picache for Successfully Defending Their Dissertation08.24.2020 Dr. James Poland dissertation (defended February 2020) is titled “Method Development towards Probing the Gut Metabolome using Reverse Phase Liquid Chromatography-Ion Mobility-Mass Spectrometry.” Dr. Jackie Picache’s dissertation (defended August 2020) is titled “Strategies for Improving Multiomic Metabolite Identifications Using Compound Libraries, Machine Learning, and Structural Mass Spectrometry”. |